
Abstract
Genomic analyses of Drosophila species suggest that the X chromosome presents an unfavourable environment for the expression of genes in the male germline. A previous study in D. melanogaster used a reporter gene driven by a testis-specific promoter to show that expression was greatly reduced when the gene was inserted onto the X chromosome as compared with the autosomes. However, a limitation of this study was that only the expression regulated by a single, autosomal-derived promoter was investigated. To test for an increase in expression associated with ‘escaping’ the X chromosome, we analysed reporter gene expression driven by the promoters of three X-linked, testis-expressed genes (CG10920, CG12681 and CG1314) that were inserted randomly throughout the D. melanogaster genome. In all cases, insertions on the autosomes showed significantly higher expression than those on the X chromosome. Thus, even genes whose regulation has adapted to the X-chromosomal environment show increased male germline expression when relocated to an autosome. Our results provide direct experimental evidence for the suppression of X-linked gene expression in the Drosophila male germline that is independent of gene dose.
Similar content being viewed by others


Introduction
Although the X chromosome is nearly identical to the autosomes in its gene density and organisation, genes residing on the X chromosome experience a very different environment than autosomal genes in terms of natural selection and gene expression (Vicoso and Charlesworth, 2006). The ploidy of the X chromosome differs between the sexes, with females having two copies and males having only one, and this has several important consequences. First, over the course of its evolution, the X chromosome is present twice as often in females as in males. This may lead to ‘feminization’ of the X chromosome and the accumulation of sexually antagonistic mutations with dominant female-beneficial effects (Rice, 1984; Charlesworth et al., 1987; Sturgill et al., 2007). Second, the hemizygosity of the X chromosome in males allows selection to be more effective on X-linked than autosomal recessive mutations. This may result in faster adaptive evolution at X-linked loci and the accumulation of sexually antagonistic mutations with recessive male-beneficial effects on the X chromosome (Rice, 1984; Charlesworth et al., 1987; Vicoso and Charlesworth, 2006; Baines et al., 2008). Third, the difference in copy number between the X chromosome and the autosomes can create an imbalance in expression, which is often overcome by mechanisms of dosage compensation (Mank, 2009; Vicoso and Bachtrog, 2009).
A fourth difference between the X chromosome and the autosomes is that the X chromosome appears to be transcriptionally silenced in the male germline, a phenomenon also known as meiotic sex chromosome inactivation (MSCI). MSCI was proposed on the basis of cytological and genetic observations (Lifschytz and Lindsley, 1972). For example, precocious condensation of the X chromosome in spermatocytes has been reported in various species, including Drosophila melanogaster (for example, Henking, 1891; Cooper, 1951). In D. pseudoobscura, this condensation has been observed for the ancestral X chromosome but not for the neo-X, which was derived recently from an autosome (Lifschytz and Lindsley, 1972). However, cytological studies have produced conflicting results (McKee and Handel, 1993) and their support for MSCI should be considered tentative (Cooper, 1951).
More recent studies have provided empirical support for MSCI in a variety of species, including mammals (Richler et al., 1992; Handel et al., 1994; Turner, 2007), Caenorhabditis elegans (Fong et al., 2002; Kelly et al., 2002) and D. melanogaster (Hense et al., 2007; Vibranovski et al., 2009a). However, there is currently debate regarding the extent of X-linked germline expression silencing in Drosophila and whether it occurs through the same mechanism described as MSCI in other taxa. Vibranovski et al. (2009a) performed a microarray analysis of gene expression in dissected regions of testes that were enriched for mitotic and meiotic cells and found a significant excess of genes whose expression was downregulated in the meiotic region, which is consistent with MSCI. In contrast, subsequent studies by Meiklejohn et al. (2011) and Mikhaylova and Nurminsky (2011) failed to find evidence for MSCI. This discrepancy has been attributed to the statistical methods that were employed, and a re-analysis of the data under the statistical framework of Vibranovski et al. (2009a) revealed a significant excess of meiotically downregulated genes in both data sets (Vibranovski et al., 2012). However, it has been pointed out that, even if the effect is significant, the X-chromosomal downregulation seen in Drosophila is much weaker than the well-known MSCI that occurs in mammals (Mikhaylova and Nurminsky, 2012).
Meiklejohn et al. (2011) also reported an absence of dosage compensation in the male germline, which results in an average 1.5-fold reduction in the expression of X-linked genes relative to autosomal genes. However, this result has been called into question, as both Meiklejohn et al. (2011) and Deng et al. (2011) found that the difference between X-linked and autosomal gene expression was greater in the testes of wild-type males than those of bag of marbles mutants, in which germ cell differentiation does not progress beyond mitosis. This suggests that at least some level of dosage compensation occurs in mitotic cells of the male germline, and suppression of X-chromosomal gene expression increases at meiosis (Deng et al., 2011, but see Meiklejohn and Presgraves, 2012).
Hense et al. (2007) showed that autosomal insertions of a transgenic construct containing the promoter of the testis-specific ocnus (ocn) gene fused to a lacZ reporter gene had significantly higher expression than X-linked insertions of the same construct. As the copy number of the reporter gene was the same for both the autosomal and X-linked insertions, these results could not be explained by a lack of dosage compensation and, thus, suggested that another mechanism functions to suppress X-linked gene expression in the male germline. However, a limitation of the Hense et al. (2007) study was that it used only a single promoter sequence that came from an autosomal gene. Thus, it is not known whether the results are relevant to other promoters and, in particular, to promoters of X-linked testis-expressed genes, which presumably have evolved to provide high expression in the male germline. In other words, the previous experiment showed that relocating an autosomal gene to the X chromosome decreased its expression but not that the relocation of an X-linked gene to an autosome increased its expression. In the present study, we demonstrate the latter using transgenic reporter genes driven by promoter sequences of three different X-linked testis-expressed genes (CG10920, CG12681 and CG1314). In all cases, we find significantly higher expression of transgenes inserted on the autosomes relative to those inserted on the X chromosome. Our results provide direct experimental evidence for the general, dosage-independent transcriptional suppression of X-linked genes during spermatogenesis.
Materials and methods
Transformation vector construction
Putative promoter sequences of three X-linked genes (CG10920, CG12681 and CG1314) were PCR-amplified from genomic DNA of the Canton S strain of D. melanogaster. The CG10920 promoter corresponds to bases 7 748 179–7 748 758 of the X chromosome (FlyBase release 5.50). The CG12681 promoter corresponds to bases 4 769 051–4 769 815, and the CG1314 promoter corresponds to bases 20 740 370–20 740 877. All of the amplified sequences lie just upstream of their respective coding sequences and end at base –28 (CG10920), –10 (CG12681) and –4 relative to the start codon.
The amplified PCR products were cloned directly into the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA, USA). The identity and orientation of the PCR fragments were confirmed by restriction analysis. A 3.6-kb NotI fragment of the pCMV-SPORT-βgal plasmid (Invitrogen) containing the Escherichia coli lacZ-coding region was cloned into the NotI site of the promoter-containing plasmid. Afterward, we performed restriction analysis to ensure that both the promoter and the lacZ-coding sequence were in the same transcriptional orientation. In a final step, an SpeI/XbaI fragment containing both the promoter and the lacZ-coding sequence was ligated into the pP[wFl] transformation vector (Siegal and Hartl, 1996). This vector is derived from the P transposable element and contains the D. melanogaster white (w; here in the form of mini-white) gene as a selectable marker.
Germline transformation
All transformation vectors were purified with the QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany) and were eluted from the column with injection buffer (0.1 mM sodium phosphate pH 6.8, 5 mM KCl). Vector DNA at a concentration of 200 ng μl−1 was used for microinjection of early-stage embryos of the strain yw; Δ2-3, Sb/TM6. The stable genomic P-element transposase Δ2-3 on the third chromosome served as the source of transposase. After microinjection, all surviving flies were crossed to a yw strain to remove the transposase source and establish stable lines. The offsprings of this cross were screened for red eye colour (imparted by the wild-type w+ gene of the vector), which was the diagnostic for stable germline transformants. Additional mobilisations of transgenes to and from the X chromosome were carried out through genetic crosses with the Δ2-3 transposing-containing stock as described previously (Hense et al., 2007).
The chromosomal location of each transgene (X or autosome) was mapped initially by genetic crosses. Transformed males were mated to yw females, and inheritance of the w+ marker was observed in the next generation. Transformed lines with X-linked insertions were identified as those producing only daughters that carry the w+ allele. Subsequently, the exact chromosomal position of each transgene insertion was determined using inverse PCR (Bellen et al., 2004). Briefly, genomic DNA was digested with HpaII or Hinp1I, and the resulting fragments were self-ligated with T4 DNA-Ligase (New England Biolabs, Ipswich, MA, USA). The target sequence, the inserted expression construct, was amplified with two primer pairs either Pry1 (5′-CCTTAGCATGTCCGTGGGGTTTGAAT-3′) and Pry2 (5′-CTTGCCGACGGGACCACCTTATGTTATT-3′) or Plac1 (5′-CACCCAAGGCTCTGCTCCCACAAT-3′) and Plac4 (5′-ACTGTGCGTTAGGTCCTGTTCATTGTT-3′). The resulting PCR products were sequenced using the above primers and BigDye v1.1 chemistry on an ABI 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). DNA sequences were used for a BLAST search of the D. melanogaster genome (FlyBase release 5.50) to determine the exact position of transgene insertion.
β-galactosidase assays
To avoid any confounding effects of transgene dosage on comparisons when comparing transformant flies with X-linked and autosomal insertions, all β-galactosidase assays were performed on flies heterozygous (autosomal) or hemizygous (X-linked) for the transgene insertion. These flies were generated by mating transformants to a yw stock. Offsprings were collected separated by sex shortly after eclosion and then maintained in standard food vials for 4–6 days prior to protein extraction.
For each enzymatic assay, six flies were homogenised in 150 μl of a buffer containing 0.1 M Tris-HCl, 1 mM EDTA and 7 mM 2-mercaptoethanol at pH 7.5. The homogenate was kept on ice for 15 min, then centrifuged at 12 000 g for 15 min at 4 °C. Enzymatic assays were performed using 50 μl of supernatant and 50 μl of assay buffer (200 mM sodium phosphate pH 7.4, 2 mM MgCl2, 100 mM 2-mercaptoethanol) containing 1.33 mg ml−1 o-nitro-phenyl-β-D-galactopyranoside. β-galactosidase activity was measured spectrophotometrically at a wavelength of 420 nm over a period of 45 min at 25 °C. The slope of the absorbance in relation to the incubation time was used to determine the amount of β-galactosidase and the relative expression between the autosomal and X-linked insertions. For each transformed line, β-galactosidase activity was measured for three biological replicates, each with two technical replicates.
Testis in situ hybridisations
Testes were dissected from males that were either heterozygous (autosomal) or hemizygous (X-linked) for the reporter gene insertion and were used for whole-tissue in situ hybridisations following the procedure described by Morris et al. (2009). The specific lines used for in situ hybridisation are indicated in Supplementary Table S1. The probe was prepared using specific forward (5′-CAAAACTCTCAAGCAGCA-3′) and reverse (5′-GATGTGGATTGGCGATAA-3′) primers to amplify ∼1 kb of the pCMV-SPORT-βgal plasmid, which included a portion of the lacZ-coding region as well as the T7 promoter of the vector. The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen), and an antisense RNA DIG-labelled probe was synthetised using T7 RNA polymerase (Roche, Basel, Switzerland) and DIG RNA Labeling Mix (Roche) as described by the manufacturer. Testes from autosomal and X-linked transformants were processed in parallel and a constant staining time of 1.5 h was used for all samples.
Quantitative reverse-transcription PCR (qRT-PCR)
Total RNA was extracted from flies heterozygous (or hemizygous) for the transgene insertion using Trizol (Invitrogen) and following the manufacturer’s protocol. Beginning with 5 μg of total RNA, DNaseI treatment was carried out for 1 h at room temperature. Afterwards, the RNA was reverse-transcribed using the Superscript II reverse transcriptase and random hexamer primers (Invitrogen). A custom-designed TaqMan probe (Applied Biosystems) was used to quantify relative lacZ mRNA abundance using a Bio-Rad CFX 96 real-time PCR machine (Bio-Rad, Hercules, CA, USA). As an internal reference, a probe to the ribosomal protein gene RpL32 (probe number Dm 02151827_g1) was used. Relative transcript abundance was measured as the difference in threshold cycle (ΔCt ) between the target and the reference gene. The difference in transcript abundance between lines with X-linked and autosomal transgene insertions was measured as the average difference in ΔCt among lines (ΔΔCt).
Quantiative analysis of mini-white expression
As a proxy for mini-white expression, we measured eye pigmentation in 4- to 6-day-old flies of both sexes using the approach of Majumder et al. (2009). Briefly, 20 heads were homogenised in 50 μl AEA (30% EtOH, 0.1% concentrated HCl) buffer and incubated at 22 °C for 30 min while shaking at 800 r.p.m. Afterwards, 1 μl of 0.5% H2O2 was added and the solution was centrifuged for 10 min at 10 000 g. The supernatant was used for spectrophotometrical measurement of the eye pigmentation at 480 nm. In total, we performed four replicate measurements (two biological replicates, each with two technical replicates) for each genotype and sex. In all cases, we used flies carrying only a single copy of the transgene (that is, males were either hemizygous or heterozygous and females were heterozygous for the insertion).
Results
Functional analysis of three X-linked, testis-specific promoters
To functionally test for an increase in male germline gene expression associated with escaping the X chromosome, we performed experiments using the upstream regulatory sequences of three X-linked, testis-specific genes: CG10920, CG12681 and CG1314. These genes are located in different regions of the X chromosome and were chosen because they show significantly male- and testis-biased expression (Table 1). In addition, for all three genes the McDonald–Kreitman test (McDonald and Kreitman, 1991) indicates a significant excess of amino-acid replacements between D. melanogaster and its sister species D. simulans (Baines et al., 2008), which is a hallmark of adaptive evolution.
As functional information about the regulatory sequences of CG10920, CG12681 or CG1314 was not available, we identified putative promoter sequences responsible for the testis expression of the three genes by comparative sequence analysis. Using aligned upstream sequences from D. melanogaster, D. simulans, D. yakuba, D. erecta and D. sechellia, we chose conserved regions of 580 bp (CG10920), 765 bp (CG12681) and 508 bp (CG1314) for further functional analysis (see Materials and methods).
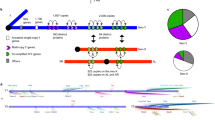
Putative promoter sequences were fused to the E. coli lacZ gene (encoding β-galactosidase) and cloned into the pP[wFl] transformation vector (Siegal and Hartl, 1996) (Figure 1). Stably transformed D. melanogaster strains were generated by embryo microinjection and subsequent genetic crosses. We recovered eight independent autosomal insertions each of the CG10920, CG12681 and CG1314 reporter gene constructs. β-galactosidase enzymatic assays indicated that all three reporter gene constructs showed highly male-biased expression (Table 2). In all cases, the difference in expression between males and females was highly significant (Mann–Whitney test, P<10−4). Additionally, we compared the β-galactosidase activity in dissected testes to that in the remaining carcass of male flies transformed with each reporter gene construct. In all cases, expression was at least 140-fold higher in the testes than in the carcass. Furthermore, in situ hybridisations indicated that there was a high level of reporter gene expression in the testis (Figure 2a). All three constructs showed lower expression in the apical tip of the testis, which is enriched for mitotic cells, than in the mid- and posterior testes, which are enriched for meiotic and post-meiotic cells, respectively (Figure 2b). This pattern was especially pronounced for the CG10920 and CG12681 constructs (Figure 2b). The CG1314 construct consistently displayed lower reporter gene expression than the other two constructs (Table 2), presumably because the CG1314 promoter fragment was a relatively weak driver of gene expression.

Reporter gene constructs. Promoter sequences of three X-linked, testis-expressed genes were fused to the E. coli lacZ reporter gene and independently inserted into the pP[wFl] transformation vector (Siegal and Hartl, 1996). This vector contains terminal repeat sequences of a Drosophila transposable element (P) and the mini-white gene as a selectable marker (eye colour). The portion of the plasmid required for replication in E. coli is labelled ‘pUC’.

In situ hybridisation of a lacZ probe in the testes. (a) Testes were dissected from males containing a single autosomal or X-linked insertion of each reporter gene construct and hybridised with a probe specific to the lacZ reporter gene. Dark blue areas indicate the presence of reporter gene mRNA. The magnification is × 200. (b) Enlargement of the testis apex from males with autosomal insertions of each reporter gene construct. The CG10920 and CG12681 show very low expression in the apical tip, which is enriched for mitotic cells. The magnification is × 600.
Comparison of X-linked and autosomal reporter gene insertions
In addition to the autosomal insertions described above, we also recovered seven, eight and nine X-linked insertions of the CG10920, CG12681 and CG1314 reporter constructs, respectively. As expected, all three constructs showed male- and testis-biased expression (Table 2). In all cases, the difference in expression between males and females was significant (Mann–Whitney test, P<10−4). Additionally, we compared the expression in the dissected testis with that in the remaining carcass of male flies transformed with each reporter gene construct. In all cases, expression was at least 12-fold higher in the testes than in the carcass.
Although the X-linked insertions of all three promoter constructs showed expression in the testis (Figure 2a), their level of expression was significantly lower than that of autosomal insertions (Figure 3). The average differences in β-galactosidase activity between autosomal and X-linked insertions were 2.8-, 3.9- and 2.9-fold for the CG10920, CG12681 and CG1314 reporter constructs, respectively.

Expression of autosomal and X-linked reporter gene insertions in adult males. The upper row shows the mean β-galactosidase activity of transformants with autosomal (grey bars) and X-linked (open bars) insertions of each reporter gene construct. Each bar represents an independent insertion at a different genomic location. The lower row shows the relative expression of the lacZ gene as determined using qRT-PCR. For each construct, the expression of the lowest line is set to 1 and all other expression values are scaled accordingly. The order of the bars corresponds to that in the upper row. In all cases, autosomal expression was significantly greater than X-linked expression (Mann–Whitney test, P<0.001). Error bars indicate the s.d.
To confirm the above results at the level of transcript abundance, we performed qRT-PCR to estimate relative levels of lacZ mRNA. For all three reporter gene constructs, the lacZ transcript abundance was significantly higher for autosomal insertions than for X-linked insertions (Figure 3). The average differences in lacZ mRNA concentration between autosomal and X-linked insertions were 2.33-, 3.01- and 3.32-fold for the CG10920, CG12681 and CG1314 reporter constructs, respectively. Thus, the estimates of transcript abundance agree well with the estimates of protein abundance. Furthermore, there was a strong correlation between expression levels measured using qRT-PCR and β-galactosidase activity (CG10920: Spearman’s ρ=0.78; P<10−5; CG12681: ρ=0.82, P<10−7; CG1314: ρ=0.66, P<0.0025).
Fine-scale mapping of transgene insertions
In order to determine the local context of the transgene insertions, we performed inverse PCR to map their precise position in the genome (Bellen et al., 2004). With this method, we were able to map eight autosomal and seven X-linked insertions for the CG10920 construct, eight autosomal and eight X-linked insertions for the CG12681 construct, and eight autosomal and nine X-linked insertions for the CG1314 construct (Figure 4). Overall, we were able to precisely map 88% of the autosomal insertions and 92% of the X-linked insertions. For all constructs, the insertions were distributed throughout the euchromatin and most (63%) were associated with genes (within a 5′ untranslated region, coding region or intron; Supplementary Table S1). The remaining insertions were in intergenic regions; however, all were within 10 kb of an annotated gene. There were no significant differences in expression among transgenes inserted into different gene regions and, within gene regions, autosomal transgene expression was always greater than X-linked transgene expression. In addition, the genomic regions surrounding autosomal transgenes did not show a significant excess of genes with testis-enriched expression in comparison to the regions surrounding X-linked transgenes (Supplementary Table S2) Thus, the observed differences in expression between autosomal and X-linked transgenes cannot be explained by differences in the local context into which they are inserted.

Map of transgene insertion locations. The precise chromosomal location of each insertion was determined using inverse PCR. Each arrow indicates an insertion at a unique site. Multiple arrows at the same position do not indicate insertions at the same site but indicate insertions that are too close to each other (within 400 kb) to be distinguished on the scale of the figure.
Analysis of somatic mini-white expression
To determine the effect of X linkage on gene expression in somatic tissues, we took advantage of the fact that all of our transformation vectors contained the mini-white gene as a selectable marker (Figure 1). This gene is derived from the X-linked white gene and shows enriched expression in the eye, where the degree of pigmentation (ranging from pale yellow to dark red) serves as an indicator of mini-white expression. We performed a spectrophotometric assay to quantify the amount of red pigment in the eyes of males and females of all of our transformed lines, using flies that were either heterozygous or hemizygous for the transgene insertion (that is, the gene dose of mini-white was always one). Comparison of the expression of individual inserts between males and females revealed a general pattern of higher expression in males (Table 3). Of the 48 independent insertions, 39 showed higher expression in males (sign test, P<0.0001). This difference is mainly attributable to X-linked insertions: 23 out of 24 X-linked insertions showed higher expression in males (sign test, P<0.0001), whereas 16 out of 24 autosomal insertions showed higher expression in males (sign test, P=0.08). These results are consistent with there being dosage compensation of X-linked mini-white gene expression in somatic tissues of males.
Unlike the testis-promoter constructs, which showed significantly greater expression when inserted on autosomes, the mini-white gene showed a trend towards greater expression when inserted on the X chromosome (Table 3). The difference between X-chromosomal and autosomal expression was marginally significant in males (Mann–Whitney test, P=0.060) but not significant in females (P=0.120). These results are consistent with dosage compensation of X-linked genes in somatic tissues and indicate that reduced X-linked expression is not a general property of our transformation vector or its preferred integration sites but instead is a feature of the male germline.
Discussion
We find that the level of testis expression driven by three different X-linked promoters is significantly increased when reporter genes are relocated to the autosomes. In combination with previous experiments that showed a reduction in testis expression when an autosomal promoter was moved to the X chromosome (Hense et al., 2007; Kemkemer et al., 2011), our results demonstrate that the X chromosome presents an unfavourable environment with respect to expression in the male germline. The three X-linked promoters used in the current study do not share sequence homology with each other or with other known testis-specific regulatory elements, which suggests that either they do not have a simple, shared regulatory mechanism or that any common regulatory sequences have diverged so extensively that they cannot be detected by a homology search. The CG12681 promoter contains a 20-bp sequence that is identical to a sequence found upstream of the male- and testis-biased gene CG5732 on chromosome arm 3R (Gnad and Parsch, 2006; Chintapalli et al., 2007). This region is predicted to contain binding sites for the Even-skipped and Zerknuellt transcription factors (Messeguer et al., 2002). However, both of these transcription factors are known to function during early embryogenesis and have no known function in spermatogenesis nor do they show enriched expression in males or testis (Gnad and Parsch, 2006; Chintapalli et al., 2007).
The exact mechanism by which X-chromosomal gene expression is suppressed in the Drosophila male germline is unknown. One possibility is that a lack of dosage compensation in the male germline leads to a general reduction in the expression of X-linked genes. Whether or not dosage compensation occurs in the Drosophila male germline is currently a subject of debate (see Introduction). However, even a complete absence of dosage compensation cannot explain our observations. This is because all of the transformed flies used in the expression assays carried only a single copy of the reporter gene. Thus, the gene dose was equal in X-linked and autosomal transformants. For this reason, our experiments are conservative, as any amount of dosage compensation would be expected to increase the level of X-chromosomal gene expression relative to that of the autosomes. Indeed, we find that the mini-white gene, which is present in all of our transformation vectors, shows expression patterns consistent with dosage compensation in the somatic (eye) tissue (Table 3). This is in agreement with previous studies reporting that, in males, X-linked alcohol dehydrogenase (Adh) transgenes show higher expression than those inserted on autosomes (Laurie-Ahlberg and Stam, 1987; Parsch et al., 1997). These findings indicate that the reduced X-linked expression seen for our testis-expressed transgenes is not an artifact of the P-element vector used for transformation, as this pattern is not observed for somatically expressed transgenes.
Another possible mechanism is MSCI, the transcriptional inactivation of the X chromosome during meiosis. A microarray analysis of gene expression during different stages of spermatogenesis indicated that there is a significant excess of X-linked genes that are downregulated during the transition from mitosis to meiosis (Vibranovski et al., 2009a), which is consistent with MSCI. However, the average decline in expression between the two stages was relatively small, suggesting that a wholesale inactivation of the X chromosome does not occur. In addition, microarray and qRT-PCR data suggest that the expression of some spermatocyte-specific genes (including CG10920, CG12681, CG1314 and ocn) increases during the mitosis–meiosis transition (Vibranovski et al., 2009a; Meiklejohn et al., 2011; Mikhaylova and Nurminsky, 2011). Our reporter gene experiments also revealed that the mRNA abundance of all three promoter constructs was relatively high in the regions of the testis enriched with meiotic and post-meiotic cells. However, there was very little expression of the CG10920 and CG12681 constructs in the the apical tip of the testis, which is enriched with mitotic cells (Figure 2b). This observation has two important implications. First, it indicates that the X chromosome is not completely inactivated at meiosis. Thus, the suppression of X-linked germline expression appears to be mechanistically different from the MSCI known to occur in mammals. Second, it suggests that contamination between stages may be an important confounding factor in studies that compare expression between dissected regions of the testes (for example, Vibranovski et al., 2009a; Meiklejohn et al., 2011). This is because the signal of expression observed in dissected ‘mitotic’ samples may come primarily from contamination with meiotic cells (Vibranovski et al. 2012). On the other hand, mRNA that is transcribed in mitotic cells will persist in meiotic and post-meiotic cells and will be detected by transcriptomic and reporter gene studies. This could explain why the observed expression difference between X-linked and autosomal insertions of our reporter gene constructs (approximately threefold) is greater than the expression difference between endogenous autosomal and X-linked genes detected using high-throughput RNAsequencing (∼1.5-fold; Meiklejohn et al., 2011). As the genes used in our study show very low expression in mitotic cells, there should be less residual signal of mitotic transcription for these genes than for many endogenous genes.
An excess of gene duplication from the X chromosome to the autosomes has been observed across the Drosophila genus (Betrán et al., 2002; Meisel et al., 2009; Vibranovski et al., 2009b). This is mainly attributable to there being a significant over-representation of retroduplicate pairs in which the parental gene is X-linked and the retrogene is autosomal (Meisel et al., 2009). Furthermore, X-to-autosome retroduplicates tend to show expression in the testis (Meisel et al., 2009). It has been proposed that selection favours retroduplicate gene copies with beneficial functions in the testis that escape the X chromosome, as they can achieve higher levels of testis expression when they are autosomal (Betrán et al., 2002). Our results are consistent with this interpretation, as all of our reporter gene constructs showed higher expression in the testis when they were relocated from the X chromosome to an autosome. In the case of retrotransposition, it is typically assumed that flanking regulatory elements are not duplicated and that new regulatory sequences are acquired from the insertion site, either by recruiting pre-existing elements or by evolving them de novo (Bai et al., 2008). Thus, it is likely that the increased expression of the autosomes in the male germline makes it easier to recruit or evolve regulatory sequences that drive high expression in the testis. Although it is difficult to establish a direct link between an increase in a gene expression in the testis and an increase in male reproductive fitness, previous findings that testis-expressed genes show high rates of adaptive evolution at the protein level (Pröschel et al., 2006; Baines et al., 2008) suggest that positive selection has an important role in the evolution of genes expressed in the male germline. Similarly, positive selection has been shown to act on testis-expressed retrogenes that have relocated from the X chromosome to an autosome (Betrán and Long, 2003; Quezada-Diaz et al., 2010; Tracy et al., 2010).
Conclusion
Previous work in D. melanogaster found that a transgenic reporter gene had significantly lower expression in the testis when inserted onto the X chromosome than the autosomes (Hense et al., 2007). This result is consistent with the suppression of X-chromosomal gene expression in the male germline. However, a caveat to the previous study was that only a single promoter derived from an autosomal gene was used (Vibranovski et al., 2012). The present study shows that the suppression of X-chromosomal gene expression extends to three additional promoters derived from X-linked genes. Thus, the observed X suppression in the male germline is independent of the promoter or its chromosome of origin. The use of transgenes allows us to examine the expression of identical genes within different chromosomal contexts and to control for gene dose, both of which are not possible in genome-wide studies of endogenous gene expression. Importantly, it allows us to rule out an absence of dosage compensation in the male germline as a cause of the reduced X-linked expression. The expression patterns of the reporter genes (Figure 2) suggest that the difference in expression between the X chromosome and the autosomes is most pronounced in meiotic and post-meiotic cells. This could be caused by a mechanism similar to the MSCI that occurs in mammals. However, the high expression of all three reporter genes meiotic and post-meiotic cells suggests that if MSCI occurs in Drosophila, it is to a much lesser extent that the MSCI that occurs in mammals. Regardless of the specific molecular mechanism, our results demonstrate that X linkage limits the expression of genes in the male germline and provide experimental support for a selective process driving the excess of X-to-autosome retroduplication that has been observed across the Drosophila genus.
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad.qt652.
References
Bai Y, Casola C, Betrán E . (2008). Evolutionary origin of regulatory regions of retrogenes in Drosophila. BMC Genomics 9: 241.
Baines JF, Sawyer SA, Hartl DL, Parsch J . (2008). Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol 25: 1639–1650.
Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G et al. (2004). The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167: 761–781.
Betrán E, Long M . (2003). Dntf-2r, a young Drosophila retroposed gene with specific male expression under positive Darwinian selection. Genetics 164: 977–988.
Betrán E, Thornton K, Long M . (2002). Retroposed new genes out of the X in Drosophila. Genome Res 12: 1854–1859.
Charlesworth B, Coyne JB, Barton NH . (1987). The relative rates of evolution of sex-chromosomes and autosomes. Am Nat 130: 113–146.
Chintapalli VR, Wang J, Dow JA . (2007). Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39: 715–720.
Cooper KW . (1951). Normal spermatogenesis in Drosophila. In: Demerec M (ed) Biology of Drosophila. Hafner: New York, NY, USA. pp 1–61.
Deng X, Hiatt JB, Nguyen DK, Ercan S, Sturgill D, Hillier LW et al. (2011). Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet 43: 1179–1185.
Fong Y, Bender L, Wang W, Strome S . (2002). Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science 296: 2235–2238.
Gnad F, Parsch J . (2006). Sebida: a database for the functional and evolutionary analysis of genes with sex-biased expression. Bioinformatics 22: 2577–2579.
Handel MA, Park C, Kot M . (1994). Genetic control of sex-chromosome inactivation during male meiosis. Cytogenet Cell Genet 66: 83–88.
Henking H . (1891). Über Spermatogenese und deren spermatogenesis Beziehung zur Entwicklung bei Pyrrhocoris apterus L. Z Wiss Zool 51: 685–736.
Hense W, Baines JF, Parsch J . (2007). X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol 5: e273.
Kelly WG, Schaner CE, Dernburg AF, Lee MH, Kim SK, Villenueve LM et al. (2002). X-chromosome silencing in the germline of C. elegans. Development 129: 479–492.
Kemkemer C, Hense W, Parsch J . (2011). Fine-scale analysis of X chromosome inactivation in the male germline of Drosophila melanogaster. Mol Biol Evol 28: 1561–1563.
Laurie-Ahlberg CC, Stam LF . (1987). Use of P-element-mediated transformation to identify the molecular basis of naturally occurring variants affecting Adh expression in Drosophila melanogaster. Genetics 115: 129–140.
Lifschytz E, Lindsley DL . (1972). The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci USA 69: 182–186.
Majumder P, Roy S, Belozerov VE, Bosu D, Puppali M, Cai HN . (2009). Diverse transcription influences can be insulated by the Drosophila SF1 chromatin boundary. Nucleic Acids Res 37: 4227–4233.
Mank JE . (2009). The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet 25: 226–233.
McDonald JH, Kreitman M . (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654.
McKee BD, Handel MA . (1993). Sex chromosomes, recombination, and chromatin conformation. Chromosoma 102: 71–80.
Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC . (2011). Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol 9: e1001126.
Meiklejohn CD, Presgraves DC . (2012). Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol Evol 4: 1007–1016.
Meisel RP, Han MV, Hahn MW . (2009). A complex suite of forces drives gene traffic from Drosophila X chromosomes. Genome Biol Evol 1: 176–188.
Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM . (2002). PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18: 333–334.
Mikhaylova LM, Nurminsky DI . (2011). Lack of global meiotic sex chromosome inactivation, and paucity of tissue-specific gene expression on the Drosophila X chromosome. BMC Biol 9: 29.
Mikhaylova LM, Nurminsky DI . (2012). No severe and global X chromosome inactivation in meiotic male germline of Drosophila. BMC Biol 10: 50.
Morris CA, Benson E, White-Cooper H . (2009). Determination of gene expression patterns using in situ hybridization to Drosophila testes. Nat Protoc 4: 1807–1819.
Parsch J, Tanda S, Stephan W . (1997). Site-directed mutations reveal long-range compensatory interactions in the Adh gene of Drosophila melanogaster. Proc Natl Acad Sci USA 94: 928–933.
Pröschel M, Zhang Z, Parsch J . (2006). Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics 174: 893–900.
Quezada-Diaz JE, Muliyil T, Rio J, Betrán E . (2010). Drcd-1 related: a positively selected spermatogenesis retrogene in Drosophila. Genetica 138: 925–937.
Rice WR . (1984). Sex chromosomes and the evolution of sexual dimorphism. Evolution 38: 735–742.
Richler C, Soreq H, Wahrman J . (1992). X inactivation in mammalian testis is correlated with inactive X-specific transcription. Nat Genet 2: 192–195.
Siegal ML, Hartl DL . (1996). Transgene Coplacement and high efficiency site-specific recombination with the Cre/loxP system in Drosophila. Genetics 144: 715–726.
Smith NG, Eyre-Walker A . (2002). Adaptive protein evolution in Drosophila. Nature 415: 1022–1024.
Sturgill D, Zhang Y, Parisi M, Oliver B . (2007). Demasculinization of X chromosomes in the Drosophila genus. Nature 450: 238–241.
Tracy C, Rio J, Motiwale M, Christensen SM, Betrán E . (2010). Convergently recruited nuclear transport retrogenes are male biased in expression and evolving under positive selection in Drosophila. Genetics 184: 1067–1076.
Turner JM . (2007). Meiotic sex chromosome inactivation. Development 134: 1823–1831.
Vibranovski MD, Lopes HF, Karr TL, Long M . (2009a). Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet 5: e1000731.
Vibranovski MD, Zhang Y, Long M . (2009b). General gene movement off the X chromosome in the Drosophila genus. Genome Res 19: 897–903.
Vibranovski MD, Zhang YE, Kemkemer C, Lopes HF, Karr TL, Long M . (2012). Re-analysis of the larval testis data on meiotic sex chromosome inactivation revealed evidence for tissue-specific gene expression related to the Drosophila X chromosome. BMC Biol 10: 49.
Vicoso B, Bachtrog D . (2009). Progress and prospects toward our understanding of the evolution of dosage compensation. Chromosome Res 17: 585–602.
Vicoso B, Charlesworth B . (2006). Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet 7: 645–653.
Acknowledgements
We thank Hedwig Gebhart for technical assistance and the Munich evolutionary biology group for helpful comments and discussions. We also thank four anonymous reviewers for their constructive comments on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft grants PA 903/5 and PA 903/6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Kemkemer, C., Catalán, A. & Parsch, J. ‘Escaping’ the X chromosome leads to increased gene expression in the male germline of Drosophila melanogaster. Heredity 112, 149–155 (2014). https://doi.org/10.1038/hdy.2013.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.86
