
Abstract
The impact of landscape structure and land management on dispersal of populations of wild species inhabiting the agricultural landscape was investigated focusing on the field vole (Microtus agrestis) in three different areas in Denmark using molecular genetic markers. The main hypotheses were the following: (i) organic farms act as genetic sources and diversity reservoirs for species living in agricultural areas and (ii) gene flow and genetic structure in the agricultural landscape are influenced by the degree of landscape complexity and connectivity. A total of 443 individual voles were sampled within 2 consecutive years from two agricultural areas and one relatively undisturbed grassland area. As genetic markers, 15 polymorphic microsatellite loci (nuclear markers) and the central part of the cytochrome-b (mitochondrial sequence) were analysed for all samples. The results indicate that management (that is, organic or conventional management) was important for genetic population structure across the landscape, but that landscape structure was the main factor shaping gene flow and genetic diversity. More importantly, the presence of organically managed areas did not act as a genetic reservoir for conventional areas, instead the most important predictor of effective population size was the amount of unmanaged available habitat (core area). The relatively undisturbed natural area showed a lower level of genetic structuring and genetic diversity compared with the two agricultural areas. These findings altogether suggest that political decisions for supporting wildlife friendly land management should take into account both management and landscape structure factors.
Similar content being viewed by others

Introduction
The majority of wildlife in many first world countries is today residing in agricultural areas, rendering these areas critical for the conservation of biodiversity (for example, Tscharntke et al., 2005). With a projection of further increase and intensification in agriculture in the upcoming years (Tilman et al., 2001) and the increase in financial support being devoted to the spread of organic farming without distinction of the various management strategies (Häring and Offerman, 2005), there is a need for prioritizing the strategies that have been proven to benefit biodiversity (Fischer et al., 2008).
It is widely believed that organic farming generally improves the diversity and abundance of species in the agricultural landscape. Many authors have investigated the effect of organic farming on wild species (see review by Hole et al. (2005) and citations therein) and the reciprocal importance of biodiversity on agricultural yield (for example, Bullock et al., 2007). However, the expectation that organic farming improves biodiversity was not met for most taxonomical groups, and it has been noticed that many other factors can be relevant to wild species in an artificial landscape (Hole et al., 2005). Moreover, it has been shown that organic farms in Europe have, on an average, a more complex landscape (regarding both field size and land cover), adding a confounding factor to the studies that compare conventional and organic fields (Norton et al., 2009). Genetic techniques can be used to detect population structuring, reduced gene flow and inbreeding, well before these effects can be seen in a reduced fitness or critically diminished population size (Ne; Frankham, 1995). Moreover, the most probable causes behind a specific population structure or gene flow pattern can be determined by comparing genetic and environmental data, without the need for manipulative studies (Storfer et al., 2007). Although the effectiveness of this approach has been shown in natural settings, there are only a few studies dealing with the effects of different agricultural management systems on the genetics of animal species (for example, Sander et al., 2006).
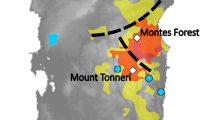
In this study, we investigate the relation between genetic diversity, isolation of populations and land management (both considering landscape connectivity and pesticide usage) in an agricultural landscape using (presumed) neutral genetic markers, microsatellites and a mitochondrial marker, cytochrome-b) and high-resolution aerial pictures (Figure 1). Mitochondrial DNA is maternally inherited and thus the results from cytochrome-b data might differ from the microsatellite data because of the strong female philopatry typical of this species (Sandell et al., 1991). To diminish the effect of random-/site-specific factors, we repeated the sampling over 2 consecutive years in two independent areas and included a relatively undisturbed natural area for comparison. This also gave the opportunity to estimate effective Ne in the areas.

Details of the two agricultural areas (Kalø and Fussingø) and the natural area (Tipperne). Sampling site FC1 lies 15 km southwest of Fussingø area. Bold lines within the sampling sites represent the trapping transects. The second letter of the sampling site name indicates the management type (C, conventional; O, organic). The position of the three areas within Denmark is shown in the bottom-right map.
We chose the field vole (Microtus agrestis) as a model organism as it is common and historically present in grassland areas and small biotopes within the agricultural landscape. Its distribution range encompasses most of Eurasia and adult vole dispersal ability—around 100 m Sandell et al., 1991)—is comparable with the fields’ dimension (making it easier to estimate landscape connectivity from a human point of view).
Moreover, field vole’s favourite habitat is represented by open grassland and it can use linear landscape features, such as hedgerows and grassy field margins, for dispersing (Yletyinen and Norrdahl, 2008). Contrary, ploughed fields are rarely crossed in the presence of other options, making them acting as barriers in much the same ways as roads or forests (Yletyinen and Norrdahl, 2008). Open grassland in the agricultural landscape is normally present either as set-aside fields, cultivated grasslands or small biotopes as hedges or ditches interspersed within cultivated fields. Consequently, from the field vole’s point of view, a cultivated landscape represents a highly fragmented habitat where the presence of hedgerows and the size of suitable patches (where new territories can be created) are fundamental for its dispersal. Finally, this species has been studied thoroughly, both genetically and ecologically, in a large part of its range (for example, Hansson, 1971; Hellborg et al., 2005), showing high genetic variability throughout (Jaarola and Searle, 2002).
Given these ecological characteristics of the field vole, we expect to see an effect of habitat fragmentation leading to a higher degree of genetic structuring (that is, a higher number of genetic clusters) in agricultural areas relative to an undisturbed natural area consisting of uniform grassland. Moreover, we expect that factors structuring the landscape such as size of the available habitat, presence and size of long-standing habitat patches, isolation of patches and presence of barriers to dispersal (for example, roads, big forests) would affect both the level of genetic differentiation and the direction and strength of gene flow of the populations. Further, we expect mortality to be higher in conventional sampling sites (because of pesticide presence and heavier managing); therefore, we hypothesize that organic fields act as genetic reservoirs within the landscape causing gene flow towards conventional fields to be higher than gene flow directed from conventional to organic fields. Finally, we expect that the amount of habitat/resources available will influence the Ne.
The main hypotheses were the following: (i) organic farms act as genetic sources and diversity reservoirs for species living in agricultural areas and (ii) gene flow and genetic structure in the agricultural landscape are influenced by the degree of landscape complexity and connectivity. They were tested by: (i) analysing differences in genetic diversity and structuring in the agricultural areas; (ii) analysing the effect of field management and other important landscape structures (for example, landscape connectivity, area of the sampling site and the amount of undisturbed habitat within each sampling site) on gene flow and genetic diversity; and (iii) analysing the effect of available habitat on Ne.
Materials and methods
Sampling and sampling sites
A total of 443 individuals of field vole (M. agrestis) were sampled in Denmark in two agricultural areas: Kalø (latitude: 56°18′00″ N longitude: 010°29′00″ E) and Fussingø (latitude: 56°23′00″ N, longitude: 009°40′00″ E) and from a relatively undisturbed natural area (Tipperne area, latitude: 55°52′00″ N, longitude: 008°13′48″ E Denmark). Samples were collected twice in each area (162 individuals in 2007 and 283 in 2008). The three areas were of comparable size, and the two agricultural areas presented both conventional and organic sampling sites (Table 1, Figure 1).
In each of the two agricultural sampling locations, different grassland patches and/or small biotopes were selected as sampling sites. Trapping transects consisted of 135 m linear transects with a trap every 15 m, and voles were captured alive and released after tipping the tail. Trapping transects were distributed in the central part of the grassland patches and, for each sample, we recorded the collection date and global positioning system coordinates together with transect information: position, sampling site, habitat type (grassland areas, small biotopes such as ditches within cultivated fields or hedgerows within fields) and management (organic or conventional, according to their certification). In Tipperne area (a continuous grassland habitat), transects were distributed evenly throughout the area (no global positioning system information was recorded for samples collected in this area). Information on samples and sampling site are summarized in Table 1 and Figure 1.
Sites characteristics and connectivity
Using high-resolution aerial pictures, the following characteristics were determined for each sampling site in Kalø and Fussingø areas: (i) area of the sampling site (m2) and core habitat area within the sampling site (m2); (ii)perimeter (both of whole sampling site and of core area); and (iii) linear distance between the centre of sampling sites. The area boundaries were drawn according to the following criteria (in order of importance): (i) barriers such as roads, water, forest border; (ii) natural division of grassland; (iii) with no clear separation, boundaries were set 100 m (average adult dispersal distance; Sandell et al., 1991) from the tip of the outermost transect. Core habitat area was defined as undisturbed area outside rotation for several years (based on the presence of shrubs and small trees identified from the aerial picture).
To estimate the ‘ecological connectivity’ of the landscape (sensu Fischer and Lindenmayer, 2007), the area surrounding the sampling sites was divided in four classes depending on the crossing possibility: (i) stable barriers (roads, water, buildings); (ii) coniferous forest (normally not used for dispersal); (iii) deciduous forest hedge, rarely used for dispersal; and (iv) everything else (fields, hedgerows and grassland), which facilitate field vole dispersal. Indices for inter-sampling site connectivity and sampling site permeability were derived from the aerial pictures taking into account the previous information. The connectivity index reflects dispersal possibility between sampling sites; if stable barriers (water bodies, roads or forests) were present between two fields a value of 0 was assigned, otherwise a value of 1 was assigned. The permeability index was defined assigning a value of 1 (barriers present on all sides), 2 (few barriers neighbouring the sampling site) or 3 (no barriers in the vicinity). Both agricultural areas were traversed by a road; therefore, the side of the road where the sampling sites were placed was also recorded. Characteristics of the different sampling sites are summarized in Figure 1 and Supplementary Information, Supplementary Table 1.
Sample storage and DNA extraction
Samples consisted of tail tips stored in saturated salt—20% dimethyl sulfoxide solution and frozen before DNA extraction. DNA was extracted using a standard cetyl trimethyl ammonium bromide buffer and proteinase-K procedure (Milligan, 1992).
Microsatellites and cytochrome-b amplification
The 15 microsatellite markers used were either developed specifically for M. agrestis: MAG6, MAG8, MAG13, MAG18, MAG21, MAG25, MAG26 (Jaarola et al., 2007) or for the congeneric species M. arvalis: Ma9, Ma25, Ma29, Ma30, Ma36, Ma54, Ma66, Ma68 (Gauffre et al., 2007). The amplification of the 15 loci was performed using two multiplex runs (markers used for first multiplex run were Ma9, Ma25, Ma66, Ma68, MAG6, MAG13, MAG18, MAG21; whereas that for the second multiplex run were Ma29, Ma54, Ma36, MAG26, MAG8, MAG25) plus a single run with Ma30 marker. Thermal profile was: 94 °C for 10 min; then 35 cycles of 45 s at 95 °C, 45 s at annealing temperature and 30 s at 72 °C, with a final extension at 72 °C for 10 min. Annealing temperatures were 52 °C for the first multiplex and the single run and 55 °C for the second multiplex run. Fragments’ lengths were analysed using an ABI 3730 automated sequencer. Microsatellite markers were typed and checked using GENEMAPPER version 4.1 (Applied Biosystems).
The central part of cytochrome-b (357 bp) was amplified using primers L15162M and H15576M (obtained from Jaarola and Searle (2002)). Thermal profile was: 94 °C for 10 min; then 30 cycles of 60 s at 95 °C, 60 s at 49 °C and 60 s at 72 °C, with a final extension at 72 °C for 8 min. Both strains of the obtained DNA fragments were sequenced (Macrogen, Seoul, Korea), and sequences were aligned and eye checked using Sequencher software version 4.2 (Gene Codes Corp., Ann Arbor, MI, USA).
Analyses of cytochrome-b data
Cytochrome-b haplotypes were determined and listed per sampling site using POPSTR software (version 1.26; HR Siegismund, personal communication). All the haplotype sequences were checked in GenBank by using BLAST (NCBI). Genetic variability was estimated as number of haplotypes, mean number of pairwise differences and gene diversity for each site using ARLEQUIN version 3.1 (Excoffier et al., 2005).
The population structure within and between the three areas was also estimated using pairwise multilocus FST(mtDNA) between sampling sites (ARLEQUIN v. 3.1, Excoffier et al., 2005).
Analyses of microsatellite data
Genetic diversity
Genetic diversity was estimated as allelic richness (FSTAT, Goudet, 2001), number of private alleles (estimated as the average of private alleles across all the loci: total number of private alleles/number of loci) and observed and expected heterozygosity (GENALEX, Peakall and Smouse, 2006). Tests for goodness-of-fit to Hardy–Weinberg expectations and linkage equilibrium were performed in FSTAT (Goudet, 2001).
Analysis of population differentiation and structure
Population structure was evaluated for the whole data set and for the three single areas using two Bayesian-based cluster analyses: STRUCTURE 2.3.3 (Pritchard et al., 2000) and GENELAND (Guillot et al., 2005) R package using different priors.
The analysis of the whole data set in STRUCTURE 2.3.3 was based on five independent runs and k(max)=10 (using 100 000/1 000 000 iterations (burnin/sampling), admixture model, correlated allele frequencies and no prior population information). STRUCTURE analysis was also performed for the three areas Fussingø, Kalø and Tipperne separately running three independent runs and k(max)=7 (using 100 000/1 000 000 iterations, admixture model, correlated allele frequencies; with no prior population information for Tipperne and with sampling sites as priors for Fussingø and Kalø). The number of clusters was determined using Structure Harvester (Earl, 2009), which relies on the method of Evanno et al (2005). The final results for the STRUCTURE analyses were visualized by clumping the separate runs using CLUMPP (Greedy algorithm with random input order and 1000 repeats, G′ statistic; Jakobsson and Rosenberg, 2007) and Distruct1.1 (Rosenberg, 2004). GENELAND analysis was performed for Kalø and Fussingø data using the admixture model and geographical coordinates (of the transects) as prior and k(max)=8 (based on three separate runs each with 50 000/200 000 iterations). The population structure within and between the three areas was also estimated using pairwise multilocus FST between sampling sites (ARLEQUIN v. 3.1, Excoffier et al., 2005).
Effect of management and landscape factors on genetic diversity
Effects of landscape factors were analysed in the two agricultural areas. For Fussingø area two data sets were considered: one including all fields and one excluding the more distant field (FC1, 15 km apart). The two data sets will be referred to as Fussingø and Fussingø without distant field. As the relatively undisturbed natural area, Tipperne, is represented by a continuum of grassland, these analyses were not applicable.
The tested environmental variables were divided in seven site-specific environmental variables: sampling site size (area), size of the core area (core area), sampling site perimeter (perimeter), core area perimeter (core perimeter), management (conventional or organic, management), percentage of perimeter usable for dispersal (defined as the percentage of perimeter free from roads and water bodies, %free perimeter), permeability index (permeability); and three pairwise environmental variables: connectivity index between sampling sites (connectivity), geographical distance and side of the road (roadside). The effect of the site-specific characteristics and the location (Fussingø and Kalø) on allelic richness, expected heterozygosity and inbreeding coefficient was tested in linear regression analyses assuming normally distributed residuals, where the most suitable linear model was selected using the Akaike information criterion (AIC) by the procedure STEP in the R software, and model assumptions were checked by plotting the residuals and from a quantile-quantile (Q-Q) plot. The significance of the single variable was then tested with an F-test using DROP1 procedure in R.
The effect of the pairwise factors (geographical distance, connectivity and roadside) on pairwise FST was tested in a simple and partial Mantel test (Mantel, 1967) using R Mantel test package with 10 000 permutations. The partial Mantel test was performed testing pairwise FST against geographical distance controlling for roadside or connectivity and vice versa.
Effect of landscape factors on gene flow
To evaluate the effect of environmental variables on the pattern of gene flow between sampling sites, BIMr (Faubet and Gaggiotti, 2008) was run on the same data sets (Kalø, Fussingø and Fussingø without distant field) used in the previous analyses. BIMr estimates recent gene flow and tries to find the best explanatory factors for the recovered pattern among the given environmental distance matrices using a linear model fit with the gene flow matrix acting as dependent variable (a null model, including none of the specified factors, is always included in the analysis). The variables included in the analysis were distance, area, core area, perimeter, core perimeter, management, %free perimeter, connectivity, permeability and roadside.
For every analysis with BIMr, five runs were performed and the best run was selected on the basis of both Dassignment and posterior probability, as suggested by Faubet et al. (2007). All runs were performed using 2 000 000/1 000 000 runs. The value of 2 000 000 was chosen as the runs converged after around 1 600 000 iterations. To decrease the uncertainty of the results, the number of environmental factors and their interactions tested with this analysis was sequentially reduced starting from the full model (all factors and all interactions) based on their posterior probabilities in the best run (that is, the factor with the lowest influence was dropped each time).
Self-assignment test for Kalø and Fussingø area was performed using GENECLASS ( Piry et al., 2004; 10 000 simulations with the method of Paetkau et al. (2004), type I error rate <0.05). GENECLASS was also used to detect first-generation migrants (using L_home/L_max likelihood; Rannala and Mountain, 1997; 10 000 simulations with the method of Paetkau et al. (2004), type I error rate <0.05). The gene flow between the sampling sites, in Fussingø without distant field and in Kalø, was estimated using BIMr. As this software contemporaneously estimates the gene flow and finds the most appropriate environmental factor(s) to explain the gene flow pattern, we used the gene flow estimates from the best run identified in the previous analysis (see paragraph above).
Estimation of Ne and effect of habitat
Ne was estimated for all sampling sites using the linkage disequilibrium method as implemented in LDNE (Waples and Do, 2008). The analysis was carried out for all sampling sites and for all available years. The correlation between the estimated Ne and core area and area of the sampling site was tested by linear regression using the linear model (y=a+bx, with y=Ne and x=core area or area) implemented in R (R Development Core Team, 2009). In case the model showed a significant correlation between variables, the robustness of the model was evaluated visually inspecting the residuals’ plots. Samples with <10 individuals were excluded from the analysis.
Results
Analyses of cytochrome-b data
In the Kalø area overall five different haplotypes were observed, whereas four were observed in the Fussingø area (Table 1; GeneBank Accession numbers: JQ619237, JQ619238, JQ619239, JQ619240, JQ619241, JQ619242, JQ619243, JQ619244, JQ619245). Tipperne area had the lowest overall diversity with only two haplotypes (H9 rare, Table 2 and Supplementary Information, Supplementary Table 1). Nevertheless, differences in haplotype numbers can be attributed most likely to an effect of sample sizes (P=0.923, exact-Fisher test). However, considering the mean number of pairwise differences and the gene diversity per population (Supplementary Information, Supplementary Figure 1), as well as the frequency distribution of haplotypes (Supplementary Information, Supplementary Table 2), the fields from Fussingø are on average more variable than Kalø’s fields. The differences between Kalø and Fussingø fields were found significant for the frequency distribution of haplotypes (P<0.0001, exact-Fisher test) and the mean number of pairwise differences (P<0.05, Student’s t-test).
The pairwise FST(mtDNA) values were significant only in few cases: FO1 and FO2 were significantly different from all others (except from each other) and FO3 differed significantly from more than half of the samples (Supplementary Information, Supplementary Table 3). The distribution of cytochrome-b FST differentiation seemed to be driven by the frequency of the most common haplotype, as the three fields with the lowest frequency for this haplotype were the most differentiated ones (Supplementary Information, Supplementary Tables 2 and 3).
Analyses of microsatellite data
Genetic diversity
Expected heterozygosity ranged between 0.671 and 0.835 (Table 1). Genetic diversity (number of private alleles, heterozygosity and allelic richness) was comparable in Fussingø and Kalø and slightly lower in Tipperne. Regarding the separate sampling sites, diversity was lowest in three organic fields in Kalø and one conventional field in Fussingø (Table 1). No significant deviation from Hardy–Weinberg expectations or from linkage equilibrium was found.
Analysis of population differentiation and structure
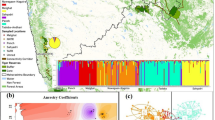
Results of the genetic structure analysis performed on the whole data set in STRUCTURE without using priors showed the presence of five clusters (Figure 2 top; log posterior probabilities graphs and barplots, for all the performed analyses, are reported in Supplementary Information, Supplementary Figures 2 and 3). One cluster was formed by all individuals from Tipperne (Figure 2 top) while the agricultural areas were divided in four clusters: one formed by the individuals from FC1 (Fussingø’s distant field), one formed by individuals from Kalø, one formed by individuals from Fussingø and one of them including the remaining individuals from both areas. The STRUCTURE analysis of the three areas taken separately and using location information as priors found two clusters in Kalø and three in Fussingø, whereas only one cluster was identified in Tipperne (Figure 2 bottom). In Kalø the organic fields were divided between two clusters, whereas the conventional field (KC1) was represented by an admixed population. In Fussingø the far conventional field (FC1) was represented by a cluster of its own, the three organic fields clustered together and the remaining two conventional fields clustered apart, although FC3 was actually represented by an admixed population (with the organic fields cluster).

Top: Bayesian clustering analysis performed in STRUCTURE 2.3.3 (Pritchard et al., 2000) for K=5 with all populations including Tipperne (T01). Bottom: Bayesian clustering analysis performed in STRUCTURE 2.3.3 (Pritchard et al., 2000) for Kalø and Fussingø areas separately (K=3 and K=2, respectively).
The population structure in the agricultural areas was investigated also using geographical coordinates as priors in GENELAND. This analysis confirmed the results obtained with STRUCTURE (Supplementary Materials, Supplementary Figure 4).
The test for population differentiation based on pairwise FST values for the microsatellite markers detected a significant differentiation between all pairs (Supplementary Materials, Supplementary Table 3), except for one pair of organic fields in Fussingø (FO2-FO3) and one in Kalø (KO3-KO4).
Effect of management and landscape factors on genetic diversity
The site-specific environmental factors analysed using linear regression analyses showed a significant positive effect of both core area and permeability on allelic richness and expected heterozygosity plus a positive effect of core perimeter on expected heterozygosity (Table 2).
Results of the analyses of the pairwise environmental factors’ effect on FST for Kalø and Fussingø areas using simple and partial Mantel tests found significant effect of geographical distance in all cases for Fussingø but not for Kalø (Table 3). Geographical distance and geographical distance given connectivity, or geographical distance given roadside were the most significant factors in Fussingø area, whereas the last one disappeared when excluding the distant field in Fussingø (Fussingø without distant field).
Effect of landscape factors on gene flow
In the BIMr analyses, four of the five runs performed for Kalø area showed a Dassignment of 0 (convergence estimator that is lowest in the best runs, Faubet et al., 2007), and thus the run with the highest posterior probability was selected. The estimated gene flow for the Kalø area could mainly be explained by core area size (Table 4) and its interactions (permeability and perimeter). The effect of core area was to increase the gene flow from the fields with smaller core area towards the fields with the larger core area. For the Fussingø and Fussingø without distant field area, none of the analysed factors could explain the estimated gene flow.
Detection of migrants and gene flow estimates
The self-assignment test performed in the agricultural areas (Supplementary Information, Supplementary Table 4) confirmed the existence of more isolated sites within the two areas. The percentage of correctly assigned individuals in Kalø was highest for the conventional and one organic field (KC1 and KO6) that were also identified by GENELAND as separate clusters. In Fussingø, the self-assignment test correctly assigned a high proportion of individuals to the sampling sites FC1, FC2 and FC3 (Supplementary Information, Supplementary Table 4). In sampling site FC2, the probability of belonging to any of the other sampling sites was rejected for 43.6% of the individuals (P<0.01, data not shown). The migrants identified by GENELAND and GENECLASS corresponded only in some cases. However, GENECLASS results confirmed the presence of migrants from the other sampling sites into KC1 in the Kalø area and the identification of dispersing individuals from FC2 into FC3 in the Fussingø area (Supplementary Information, Supplementary Table 5). These results also show that there are migrants from all the other sites into FC2 and FC3.
The gene flow estimates (Supplementary Information, Supplementary Table 5 and Supplementary Figure 4) between the sampling sites were extremely low (maximum value: 1.50 × 10−8 between FO1 and FC2) despite the conspicuous number of first-generation migrants. In Kalø, the biggest organic field (KO2) showed the highest average immigration rate, whereas the conventional field (KC1) showed the highest average emigration rate (with gene flow from KC1 to KO2). In Fussingø without distant field, the immigration/emigration rates were all of the same order of magnitude. However, FC2 had the highest emigration and FO1 had the highest immigration rate. FC2 also showed the lowest immigration rate, whereas the lowest average emigration rate was found in FC3 and FO2 (gene flow from FC2 to FO2 being lowest).
No emigrants and four possible immigrants were detected in the distant field from Fussingø (data not shown). This was probably because of chance, given the extremely low probability that a field vole disperses 15 km.
Estimation of Ne
The estimated Ne (when defined) ranged from 83 to 1960 in the Kalø area, from 3 to 277 in Fussingø and from 65 to 772 in Tipperne (Table 1). The linear regression analyses showed a clear positive correlation between core area and Ne (P=0.01, R2=0.58) and no correlation between area and Ne (Table 1). A visual inspection of the residuals’ plots demonstrated that the data fitted the model.
Discussion
Analysis of population differentiation and structure
A more defined population structure was expected in the agricultural compared with the natural area because of habitat fragmentation. From the genetic structure analysis it was shown that the three main areas and the most distant field in the Fussingø area formed four separate clusters as expected given the low dispersal ability of this species (Sandell et al., 1991; Figure 2). More interestingly, separate genetic clusters were present in the agricultural areas but not in the natural one, and the presence of the same isolated populations within the two agricultural areas were confirmed by all analyses (Figure 2). However, given that this observation was based on only one natural area it should be interpreted cautiously. In particular, n the Fussingø area, this clustering was evident also from cytochrome-b data where the haplotype distribution mirrored the genetic structure recovered by the STRUCTURE analysis (Figure 2 and Supplementary Information, Supplementary Table 2). This difference is probably linked to more limited dispersal ability and a patchier habitat distribution in the agricultural areas. A further interesting aspect, in Fussingø area, is the separate clustering of all the conventional fields, whereas organic fields form a single cluster (Figure 2).
Some investigations on small mammals (small carnivore mustelids, shrews, voles and mice) in the arable land performed on conventional farms in Denmark show that intensive agriculture leaves little space for small mammals and consequently, especially within fields, their abundance is low (Jensen, 2000; Jensen and Hansen, 2003). This indicates, together with our results, the importance of organic farming for small mammals.
Effect of management and landscape features on genetic diversity
The analyses of the effect of landscape features (Tables 2, 3, 4) on genetic diversity showed that core area is the most important factor and permeability index is the second most important factor. This proves the importance of long-standing habitat patches for the survival of a species as compared with the mere presence of suitable but fast changing habitats. It also shows that, in our case, small, low traffic roads do not have a larger impact on genetic diversity compared with other natural barriers. These findings emphasize the significance of unmanaged areas, and they are in agreement with the findings that production of seeds in uncropped areas becomes of increasing importance to the farmland’s birds and small mammals (Fuller et al., 2004). Especially, the changes from spring cereals to winter cereals and the decline in areas with stubble fields (but also the general decline in weeds and in weed seed bank) reduces the seed availability within fields (Andreasen et al., 1996; Rich and Woodruff, 1996).
Effect of landscape factors on gene flow
Results of BIMr analyses confirmed the importance of long-standing habitat patches and accessibility of the habitat (Table 4) for one of the agricultural areas (Kalø). The size of the long-standing habitat patches caused an increase in immigration, probably because of a larger amount of resources available. In the other area (Fussingø), it was not possible to identify a significant environmental factor responsible for shaping the gene flow pattern. This inability might be ascribed to the environmental differences between the two areas and underpins the importance of studying the environmental factors related to the particular area and species of interest. This is also in agreement with a study showing the importance of landscape scale factors rather than field factors for biodiversity in the agricultural landscape (Gabriel et al., 2010).
Detection of migrants and gene flow estimates
Comparing the results of the detection of first-generation migrants (dispersing individuals) with the gene flow estimates (Supplementary Information, Supplementary Table 5 and Supplementary Figure 5) is interesting. First, despite an elevated number of dispersers the gene flow is very low. These results are consistent with findings from an ecological study that has shown that dispersers are mainly young males that loose the competition for territories to the older males, and therefore have a lower probability of establishing a territory, necessary to reproduce, in another already populated area (Myllymäki, 1977). The extent of gene flow is largest in the Fussingø area compared with the Kalø area, which is also confirmed by the higher number of shared haplotypes between Fussingø sampling sites.
Second, given the present sampling design (one conventional field in Kalø and two in Fussingø), organic fields did not act as reservoirs for the conventional fields. The conventional field in Kalø and one of the two conventional fields in Fussingø without distant field had the highest emigration rates, and the same Fussingø field had the lowest immigration rate, thus the expectation on this point was not met. Rather, it seemed like individuals left the conventional fields caused by male territoriality.
In a study that correlated landscape structure to migration/dispersal factors in the field vole, Yletyinen and Norrdahl (2008) found that the size and presence of buffer zones and field margins accounted for the different use of landscape and distances moved by the individuals. Thus, these factors will certainly also influence gene flow. These observations confirm our finding that both size and accessibility of habitat patches have an important role for the field vole. All in all, there is a strong indication that landscape structure does have an effect in shaping the population diversity and gene flow patterns in the agricultural landscape, which is also confirmed by the presence of genetic structuring in the two agricultural areas.
Estimation of Ne and effect of habitat
The estimates of effective Ne in the present study (Table 1) are expected to be biased upwards (Waples and Do, 2009). The values, however, fall within the same order of magnitude of other studies calculating effective Ne for rodent species (Antolin et al., 2001; Buzan et al., 2010). In accordance with the other analyses, the estimated effective Ne showed a significantly positive correlation with the amount of unmanaged available habitat (as expected), but not with the total size of the habitat. This suggests that the amount of unmanaged habitat could be highly important in maintaining viable populations in the agricultural environment.
Conclusions
This study aimed at investigating which factors influence the pattern of genetic differentiation and gene flow in agricultural areas as compared with less disturbed areas. The expectation that a stronger habitat fragmentation, in the two agricultural areas compared with the more uniform natural area, would result in an increased genetic structuring was met, although we cannot conclude that this represents a general pattern. Further studies including more natural areas would be needed to verify this. The results also indicate that field management might have an effect on the genetic structure, which is in accordance with ecological studies (Jensen, 2000, Jensen and Hansen, 2003). However, the most important factor in shaping genetic diversity and gene flow was the size of long-standing habitat patches, followed by the accessibility of the sampling site. In agreement with other studies on bank voles (Gerlach and Musolf, 2000), no effect of road presence on the genetic diversity and gene flow was found.
Moreover, from the genetic structure and gene flow analyses, it is clear that organic fields did not act as reservoirs of genetic diversity for conventional fields, and that the latter, in both areas, were well differentiated from the more uniform clusters of organic fields. This structuring in the conventional fields might be caused by intensive genetic drift, although a bottleneck could not be detected. Thus, the presence of organic fields cannot counteract possible negative effects (such as reductions in abundance of field voles) of the presence of conventional fields. These findings are in good agreement with an ecological study, performed in England, where it was shown that the benefits of organic farming are evident only at the landscape scale and not at the field scale (Gabriel et al., 2010), and that many species might be more influenced by the landscape structure and composition rather than organic or conventional management. All in all, this study showed that although management might be an important factor, switching to organic farming is not enough to develop a wildlife friendly cultivation system, as the quality and quantity of the habitat and the habitat connectivity might be of greater importance.
Data archiving
Sequence data have been submitted to GenBank Accession numbers: JQ619237, JQ619238, JQ619239, JQ619240, JQ619241, JQ619242, JQ619243, JQ619244, JQ619245.
Genotype data deposited in the Dryad repository: doi:10.5061/dryad.3fs0q.
References
Andreasen C, Stryhn H, Streibig JC . (1996). Decline of the flora in Danish arable fields. J Appl Ecol 33: 619–626.
Antolin MF, Horne BV, Holloway AK, Roach JL, Berger JMD, Weeks JRD . (2001). Effective population size and genetic structure of a Piute ground squirrel (Spermophilus mollis) population. Canadian J Zool 79: 26–34.
Buzan EV, Krystufek B, Bryja J . (2010). Microsatellite markers confirm extensive population fragmentation of the endangered Balkan palaeoendemic Martino’s vole (Dinaromys bogdanovi). Conserv Gen 11: 1783–1794.
Bullock JM, Pywell RF, Walker KJ . (2007). Long-term enhancement of agricultural production by restoration of biodiversity. J Appl Ecol 44: 6–12.
Earl DA . (2009) Structure Harvester v0.3. Available at http://users.soe.ucsc.edu/∼dearl/software/struct_harvest/.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.1): an integrated software package for population genetics data analysis. Evol Bioinf Online 1: 47.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Faubet P, Gaggiotti OE . (2008). A new Bayesian method to identify the environmental factors that influence recent migration. Genetics 178: 1491–1504.
Faubet P, Waples RS, Gaggiotti OE . (2007). Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16: 1149–1166.
Fischer J, Brosi B, Daily GC, Ehrlich PR, Goldman R, Goldstein J et al. (2008). Should agricultural policies encourage land sparing or wildlife-friendly farming? Front Ecol Environ 6: 380–385.
Fischer J, Lindenmayer DB . (2007). Landscape modification and habitat fragmentation: a synthesis. Global Ecol Biogeogr 16: 265–280.
Frankham R . (1995). Conservation genetics. Annu Rev Genet 29: 305–327.
Fuller RJ, Hinsley SA, Swetnam RD . (2004). The relevance of non-farmland habitats, uncropped areas and habitat diversity to the conservation of farmland birds. Ibis 146: 22–31.
Gabriel D, Sait SM, Hodgson JA, Schmutz U, Kunin WE, Benton TG . (2010). Scale matters: the impact of organic farming on biodiversity at different spatial scales. Ecol Lett 13: 858–869.
Gauffre B, Galan M, Bretagnolle V, Cosson JF . (2007). Polymorphic microsatellite loci and PCR multiplexing in the common vole, Microtus arvalis. Mol Ecol Notes 7: 830–832.
Gerlach G, Musolf K . (2000). Fragmentation of landscape as a cause for genetic subdivision in bank vole. Conserv Biol 14: 1066–1074.
Goudet J . (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Institute of Ecology, University of Lausanne: Lausanne, Switzerland.
Guillot G, Mortier F, Estoup A . (2005). Geneland: a computer package for landscape genetics. Mol Ecol Notes 5: 712–715.
Hansson L . (1971). Small rodent food, feeding and population dynamics: a comparison between granivorous and herbivorous species in Scandinavia. Oikos 22: 183–198.
Häring AM, Offerman F . (2005) Impact of the EU Common Agricultural Policy on organic in comparison to conventional farms. Paper presented at the conference “Researching Sustainable Systems – International Scientific Conference on Organic Agriculture” 21-23 September 2005. Adelaide, Australia.
Hellborg L, Guenduez I, Jaarola M . (2005). Analysis of sex-linked sequences supports a new mammal species in Europe. Mol Ecol 14: 2025–2031.
Hole DG, Perkins AJ, Wilson JD, Alexander IH, Grice PV, Evans AD . (2005). Does organic farming benefit biodiversity? Biol Conserv 122: 113–130.
Jaarola M, Ratkiewicz M, Ashford RT, Brunhoff C, Borkowska A . (2007). Isolation and characterization of polymorphic microsatellite loci in the field vole, Microtus agrestis, and their cross-utility in the common vole, Microtus arvalis. Mol Ecol Notes 7: 1029–1031.
Jaarola M, Searle JB . (2002). Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol Ecol 11: 2613–2621.
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Jensen TS . (2000) In: De små pattedyr og de alt for store landskaber – Naturkvalitet i terrestriske økosystemer. I: “Aktører i landskabet” Møller PG, Holm P, Rasmussen L (eds). pp 225–235.
Jensen TS, Hansen TS . (2003). Biodiversitet og biotopfordeling af småpattedyr i det åbne land. Flora og Fauna 109: 9–21.
Mantel N . (1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
Milligan B . (1992). Plant DNA isolation. In: Hoelzel AR (eds). Molecular genetic analysis of populations: a practical approach. IRL Press: Oxford, UK. pp 59–88.
Myllymäki A . (1977). Intraspecific competition and home range dynamics in the field vole Microtus agrestis. Oikos 29: 553–569.
Norton L, Johnson P, Joys A, Stuart R, Chamberlain D, Feber R et al. (2009). Consequences of organic and non-organic farming practices for field, farm and landscape complexity. Agric Ecosyst Environ 129: 221–227.
Peakall ROD, Smouse PE . (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Paetkau D, Slade R, Burden M, Estoup A . (2004). Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13: 55–65.
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A . (2004). GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95: 536–539.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–945.
R Development Core Team. (2009) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org.
Rannala B, Mountain JL . (1997). Detecting immigration by using multilocus genotypes. P Natl Acad Sci USA 94: 9197–9201.
Rich TCG, Woodruff ER . (1996). Changes in the vascular plant floras of England and Scotland between 1930-1960 and 1987-1988: the BSBI monitoring Scheme. Biol Conserv 75: 217–229.
Rosenberg NA . (2004). Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Sandell M, Agrell J, Erlinge S, Nelson J . (1991). Adult philopatry and dispersal in the field vole Microtus agrestis. Oecologia 86: 153–158.
Sander AC, Purtauf T, Wolters V et al. (2006). Landscape genetics of the widespread ground-beetle Carabus auratus in an agricultural region. Basic Appl Ecol 7: 555–564.
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF et al. (2007). Putting the ‘landscape’ in landscape genetics. Heredity 98: 128–142.
Tilman D, Fargione J, Wolff B, D'Antonio C, Dobson A, Howarth R et al. (2001). Forecasting agriculturally driven global environmental change. Science 292: 281–284.
Tscharntke T, Klein AM, Kruess A, Steffan-Dewenter I, Thies C . (2005). Landscape perspectives on agricultural intensification and biodiversity-ecosystem service management. Ecol Lett 8: 857–874.
Waples RS, Do C . (2008). LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Notes 8: 753–756.
Waples RS, Do C . (2009). Linkage disequilibrium estimates of contemporary N e using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3: 244–262.
Yletyinen S, Norrdahl K . (2008). Habitat use of field voles (Microtus agrestis) in wide and narrow buffer zones. Agric Ecosyst Environ 123: 194–200.
Acknowledgements
We thank all the people involved in the REFUGIA project for helpful discussion. We specially thank Tine Susi Hansen who performed the sampling. Research in Organic Food and Farming, International Research Co-operation and Organic Integrity (DARCOF III 2005–2010) are thanked for financial support. This work was funded by the Ministry of Food, Agriculture and Fisheries under the Finance and Appropriation Act, Sections 24.33.02.10. Part of the Ph.D. project, to which this project belongs, was funded by AGSoS (Aarhus Graduate School of Science).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Marchi, C., Andersen, L., Damgaard, C. et al. Gene flow and population structure of a common agricultural wild species (Microtus agrestis) under different land management regimes. Heredity 111, 486–494 (2013). https://doi.org/10.1038/hdy.2013.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.70
