
Abstract
Domestic chickens (Gallus gallus domesticus) fulfill various roles ranging from food and entertainment to religion and ornamentation. To survey its genetic diversity and trace the history of domestication, we investigated a total of 4938 mitochondrial DNA (mtDNA) fragments including 2843 previously published and 2095 de novo units from 2044 domestic chickens and 51 red junglefowl (Gallus gallus). To obtain the highest possible level of molecular resolution, 50 representative samples were further selected for total mtDNA genome sequencing. A fine-gained mtDNA phylogeny was investigated by defining haplogroups A–I and W–Z. Common haplogroups A–G were shared by domestic chickens and red junglefowl. Rare haplogroups H–I and W–Z were specific to domestic chickens and red junglefowl, respectively. We re-evaluated the global mtDNA profiles of chickens. The geographic distribution for each of major haplogroups was examined. Our results revealed new complexities of history in chicken domestication because in the phylogeny lineages from the red junglefowl were mingled with those of the domestic chickens. Several local domestication events in South Asia, Southwest China and Southeast Asia were identified. The assessment of chicken mtDNA data also facilitated our understanding about the Austronesian settlement in the Pacific.
Similar content being viewed by others
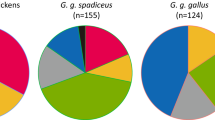


Introduction
Being the most extensively distributed of the poultries, the domestic chicken (Gallus gallus domesticus) provides humans with a stable sources of protein, including both meat and eggs (FAO, 2007). The chicken has a long history of anthropomorphic usage in Southeast and East Asia, where it has been bred for entertainment (cockfight) and ornamentation (Macdonald and Blench, 2000). The domestic chicken also serves as an important model animal in biomedical research (Wu and Kaiser, 2011). Although humans derive much benefit from this poultry, its history of domestication remains open to debate.
Since the times of Charles Darwin (Darwin, 1896), the origin and domestication of chickens has attracted wide interest from multiple disciplines (West and Zhou, 1988; Crawford, 1990). A massive amount of research has focused on reconstructing the matrilineal history of domestic chickens using mitochondrial DNA (mtDNA) sequence data. These efforts document that the red junglefowl (Gallus gallus) is the primary wild ancestor of the domestic chicken (Fumihito et al., 1994, 1996). The multiple, independent domestication events in southern China, South Asia and Southeast Asia involve several matrilines (Liu et al., 2006; Kanginakudru et al., 2008).
Most studies of chicken mtDNA rely on sequences of (partial) control region (CR; D-loop: nucleotide position 1–1232; NC_007235). The relatively small size of CR limits the resolution of the mtDNA phylogeny. Because the mutation rate in this region is higher than that in coding regions, high levels of recurrent mutations can blur the structure of the matrilineal genealogy. Recent, fine-gained analyses have used the complete mtDNA genome to reconstruct the history of animal domestication, such as in cattle (Achilli et al., 2008, 2009; Bonfiglio et al., 2010), dogs (Pang et al., 2009), horses (Achilli et al., 2012) and pigs (Wu et al., 2007). These updated phylogenies provide new insights into the origins and history of domestication.
In the present study, we selected and sequenced 50 complete mitochondrial genomes from >2000 samples of domestic chickens and red junglefowl from China, India, Laos and Myanmar. Incorporating 11 published genomes, we reconstructed the matrilineal genealogy of the chicken to further refine the tree and better document the history of domestication.
Materials and methods
Samples
We collected samples consisting of 2044 domestic chickens and 51 red junglefowl from China, India and Southeast Asia (see Supplementary dataset 1). A total of 2874 published mtDNA sequences from domestic chickens (n=2719) and red junglefowl (n=155) were retrieved from either GenBank or published literature (see Supplementary dataset 1). For the unpublished sequences, we confirmed the source and quality of sequencing. Twenty-six sequences (AB098640, AY588607, DQ834510, EF414962–EF414970, EF586879–EF586882, EU194446, EU329407, EU329413, EU367396, EU847802–EU847804, EU847806–EU847807 and FJ619040) were suggested to have sequencing errors, such as extensive indels and ambiguous sites. Four sequences (AB007734, AB007749, AB009445 and AB009447) had unknown sources, and AB368439 from ancient DNA was too short to be informative. Therefore, we excluded these 31 sequences from further analyses. In total, we obtained 4938 mtDNA sequences from 4732 domestic chickens and 206 red junglefowl (see Supplementary dataset 1).
DNA extraction, amplification and sequencing
Genomic DNA was extracted from whole blood, muscle or feather by the standard phenol/chloroform method. The amplification and sequencing of chicken CR followed protocols described in Supplementary dataset 2. An unrooted neighbour-joining network of CR sequences from 3676 chickens and 148 junglefowl was constructed by using MEGA 4.0 (Tamura et al., 2007). We used the network to select 50 samples for whole mtDNA sequencing that represented 48 haplotypes and most major groups (Figure 1). The amplification and sequencing protocols were described in Supplementary dataset 2.

The samples selected for mtDNA genome sequencing were based on the neighbour-joining network of 409 haplotypes. MtDNAs with haplotypes selected for complete sequencing are indicated with circles. Numbers in the circles correspond to the sequences in Figure 2 and Supplementary dataset 3. Numbers in red and black refer to the red junglefowl and domestic chickens, respectively. The geographic sources of the samples are represented by different colors. The discordances in the phylogeny between based on CR and complete mtDNA genome are noted by the red ellipse with dash line.
Sequence analysis
Sequences of both CR and the complete mtDNA genome were edited and aligned using DNASTAR software (DNASTAR Inc., Madison, WI, USA). The previous reference sequence NC_001323 (Desjardins and Morais, 1990) likely had sequencing errors, including excessive indels, and it was abandoned (Figure 2). Variants in the mtDNA sequences were scored relative to the new reference sequence NC_007235 (Nishibori et al., 2005). The classification of the variants of each chicken mtDNA genome was performed with mtDNA-GeneSyn v.1.0 (Pereira et al., 2009).

The haplogroup classification tree of 61 complete mtDNA sequences (a, b). The nucleotide positions in the sequences were scored relative to the reference sequence NC_007235 which is noted by the dashes. Transitions are shown on the branches and transversions are further annotated by adding suffixes. Deletions and insertions are indicated by ‘d’ and ‘+’, respectively. Amino-acid replacements are in red and marked by a single-letter code, whereas synonymous replacements are in blue. Changes in ribosomal RNA genes are denoted by ‘r’. The exclamation mark ‘!’ designates back mutation and recurrent variants are underlined. ‘R’ and ‘Y’ specify the heteroplasmic status of A/G and C/T at a certain site, respectively. All heteroplasmic variants are excluded in the analyses and are marked in italics. ‘0’ represents the previous reference sequence NC_001323 which was excluded in subsequent analyses because of potential sequence errors. Labels of sequences are in accordance with Figure 1. (c) The geographic origin of samples as shown by different colors.
Genealogy construction and data analysis
The phylogeny of 61 mtDNA genomes (50 de novo and 11 from the literature; see Supplementary dataset 3) was constructed based on the parsimony-like method as described elsewhere (Wu et al., 2007; van Oven and Kayser, 2009). In detail, variants of each mtDNA genome scored relative to NC_001323 (see Supplementary dataset 3) were either synapomorphic or autapomorphic. We mapped the mutations on each branch of the tree. Most chicken mtDNA haplogroups were defined with the diagnostic motifs (a string of characteristic mutations shared by the members) in both coding and CRs. We attempted to assign each of the CR sequences to specific haplogroups following the strategy of mutational motif search and (near-) matching (Wu et al., 2007). To test the assignment results, we screened certain coding region diagnostic motifs in 338 samples. The average sequence divergence (Rho) of the haplotypes to their most recent common ancestor (Forster et al., 1996), accompanied by a heuristic estimate of the standard error (Sigma) (Saillard et al., 2000), was calculated based on complete mtDNA and synonymous mutations alone, respectively. The synonymous mutations were suggested to be less influenced by mutation saturation and selection (Kivisild et al., 2006; Soares et al., 2009).
Results
Phylogeny of chickens based on mtDNA genomes
The chicken matrilineal phylogeny (genealogy) was reconstructed with 61 mtDNA genomes of which 50 were newly generated (Figure 2; see Supplementary dataset 3). For convenience, the nomenclature for haplogroups A–G were updated from our previous study based on CR (Liu et al., 2006), and in part because they were already used frequently. The nomenclature conformed to the rules for the human mtDNA phylogeny (van Oven and Kayser, 2009; Behar et al., 2012) for the newly defined haplogroups (that is, H, I and W–Z), subhaplogroups (for example, C1, C2 and C3) and macro-haplogroups (for example, ABZY and CD). An example of the nested hierarchy was as follows: C1→C→CD. In some cases, we labeled haplogroups (for example, H, I) and subhaplogroups (for example, C2, D2) only when supported by one sequence. The aim was to facilitate the designation of haplogroups in this study. We expected the definitions to change upon adding future sequences.
Haplogroups A–G previously defined only by CR information, were regarded as being sufficiently supported by the complete sequence data. These haplogroups were defined by diagnostic mutational motifs shared by the descendants (Figure 2). The new reference sequence NC_007235 belonged to haplogroup B. The nested hierarchy of haplogroups A–G was clarified by the macro-haplogroups AB, CD and EFG. The newly defined haplogroups (that is, H, I and W–Z) represented early splits from macro-haplogroups AB, CD and EFG. Discordances between the CR and complete mtDNA sequence data were noted. Sequences 12 and 18–20, which previously fell in clade D (Figure 1), were assigned into haplogroups Y and C, respectively (Figure 2). Sequence 60, which previously represented a basal branch in clade C (Figure 1), defined the new haplogroup X (Figure 2).
Global mtDNA profiles of chickens
The diagnostic mutational motifs in both coding and CRs for most haplogroups were characterized in the updated mtDNA phylogeny (Figure 3; see Supplementary dataset 4). In terms of motif searches, 99.9% (4727/4732) of the CR sequences in domestic chickens were allocated into haplogroups A–I. Additional sequencing of certain CR variants supported the assignments (see Supplementary dataset 1). Five published CR sequences remained unclassified. Haplogroups D and E1 were not directly defined by mutations in CR. Thus, the CR motifs relied on definitions for both the higher units (CD and E) and the sister groups (C and E2–E3). For convenience, we tentatively assigned all sequences within macro-haplogroup CD into haplogroup D while excluding those belonging to haplogroup C (Figure 3). Similarly, all sequences within haplogroup E were allocated into haplogroup E1, except for subhaplogroups E2 and E3. Haplogroups D and E1 defined by CR sequences in Figure 3 were potentially paraphyletic, and they could be called ‘paragroups.’

The hierarchical phylogenetic relationships and distributions of the haplogroups observed in global domestic chickens. The diagnostic mutational motifs for each of the haplogroups are shown on the branches. ‘Un’ means the unclassified sequences within this phylogeny. The sample sizes are in parentheses. The regions of China follow the traditional definitions (see Supplementary dataset 1).
In the re-evaluated, global mtDNA profiles of the chicken (Figure 3), haplogroup E1 was the most widely distributed matrilineal lineage. It occurred in all geographically defined populations. Its sister haplogroups E2 and E3 were mainly restricted to South Asia. Haplogroups A and B were widely distributed but not detected in Africa. Haplogroup D occurred in Africa, South Asia, Southeast Asia and East Asia. Its frequency of occurrence peaked in the Pacific Islands (∼77%). Haplogroups C, F and G were mainly distributed in East Asia, Southeast Asia and South Asia. The maternal lineages of commercial lines consisted of the three most common haplogroups A, B and E1. Haplogroups H and I were very rare being found only in domestic chickens from East Asia and South Asia, respectively. Because of the extensive sampling in East Asia (n=2732), we also assessed the mtDNA profiles of chickens from the nine subregions (Figure 3). Haplogroups A, B, C1 and E1 were distributed largely across all of East Asia. Haplogroups F and G were mainly concentrated in Southwest China. Rare haplogroup H was found in Southwest China and Japan.
As for the red junglefowl, 76.2% (157/206) of the CR haplotypes were assigned to haplogroups in this genealogy (Figure 4; see Supplementary dataset 1). Apart from the common haplogroups A–G, the wild fowl harbored haplogroups W–Z, which were not detected in domestic chickens. Of the remaining sequences (49/206) not classified in the genealogy, 28 haplotypes from India and Indonesia (for example, ‘outgroups’ in Figure 1) had too many variants to be assigned; variation included many transversions (see Supplementary dataset 1). This suggested that they were remotely related to the other chickens. It was also possible that these lineages likely originated from other wild fowl such as G. sonneratii (Eriksson et al., 2008) and G. varius (Hutagalung, 2000) through hybridizations, although comprehensive surveys for the wild fowl were needed to test these possibilities. The other 21 sequences from Vietnam and Indonesia had mutational motifs not recognized in the sequenced mitochondrial genomes; these samples were not available to us.

mtDNA haplogroup profile for 206 red junglefowl.
Molecular divergence
No suitable calibration points were available for the chicken phylogeny, neither from the fossil record nor from ancient DNA data. Consequently, we could not calculate reliable dates of divergence for the haplogroups. We calculated molecular divergences by employing the Rho method to indirectly estimate ages (Table 1). Haplogroups/subhaplogroups B, C1, E1 and F were characterized by very similar, average amounts of sequence divergences based on both the complete mtDNA and synonymous mutations only. The estimated divergences for haplogroups A and D1 were slightly larger than those for haplogroups B, C1, E1 and F, although they overlapped when considering the 95% confidence intervals (Rho±2Sigma). For haplogroup G, discordance was observed between estimates based on entire mtDNA genome and synonymous mutations. Haplogroup C1 existed only in the domestic chicken, a pattern also supported by analyses of the CR data, although it might have occurred in red junglefowl not sampled in the current study. When the estimated age of haplogroup C1 was considered as a calibration point for chicken domestication, differentiation for haplogroups A, B, E1, F and G likely represented expansion after domestication of the chicken.
Discussion
The mtDNA phylogeny extends our understanding about the matrilineal history of chickens. Lineages of common haplogroups/subhaplogroups A, B, D1, E1, E3, F and G occur in both domestic chickens and red junglefowl. Two phenomena may explain this pattern. First, the differentiation of each of the haplogroups/subhaplogroups predates the domestication of chicken. In this case, domestication is a recent event. The earliest ‘chicken’ remains (bones) are from the Cishan site (∼6000 B.C.), North China (West and Zhou, 1988). However, it is still unclear whether the remains belonged to the domestic chicken or the wild fowl (Yuan, 2010). Further, the appearance of domestic chicken dates to at least 3600 years old in China (Yuan, 2010). In South Asia, domestic chicken bones are known from the Harappa and Mohenjo-Daro sites in Pakistan from around 2500–2100 B.C. (Zeuner, 1963). Thus, the domestication of chickens might be a recent event as compared with other domestic mammals (for example, dog, cattle, sheep, pig), which predate the Mid-Holocene. Second, domestic chickens and wild junglefowl may have experienced substantial gene flow and genetic admixture following domestication of the chicken. Indeed, hybridization has been observed in Yunnan, China (Chang, 2009) and inferred in northern Vietnam (Berthouly et al., 2009). In extreme cases, some domestic chickens might have become feral with their descendants living as wild fowl. These situations are possible in the places (for example, Southeast Asia, Yunnan and Northeast India) where domestic chickens and wild fowl coexist, especially when the management of chickens is poor. The above possibilities need not be taken as alternatives. Both may play roles in the history of domestication.
The extensive sampling results in the identification of several localized mtDNA lineages. Several lineages reveal new details about chicken domestication. Haplogroups E2 and E3 occur mainly in South Asian domestic chickens and red junglefowl (see Supplementary dataset 1). This discovery provides genetic evidence for an independent domestication event in South Asia. As for rare haplogroup I, its restricted distribution suggests another localized domestication event in Northeast India. The situation for haplogroup H is puzzling. This haplogroup occurs in Southwest China (Figure 3). Several CR sequences from ‘Shamo’ Japanese gamecocks raised in Okinawa (Komiyama et al., 2003; Oka et al., 2007) show the same mutational motif and they are allocated into this haplogroup (see Supplementary dataset 1). The existence of cryptic genetic links between Southwest China and Okinawa supports the introduction of the Shamo breed from Southeast Asia (Komiyama et al., 2003; Oka et al., 2007), which neighbors with Yunnan, Southwest China. Thus, haplogroup H suggests yet another localized domestication, this one in Southwest China and neighboring Southeast Asia.
Because the chicken is easily carried during human dispersal and migration, analyses of chicken mtDNA data can contribute to the Holocene history of humans. One of the best-documented examples is the settlement of the Pacific by Austronesian speakers (Hurles et al., 2003; Patrick, 2010). Some archeological and linguistic studies have suggested that this dispersal originated from Taiwan (that is, the Out-of-Taiwan hypothesis; Diamond, 2000). The chicken is proposed to be bound to this dispersal (Bellwood, 1976; Storey et al., 2008; Dancause et al., 2011) and, thus, we ask: can the Pacific chicken be traced back to Taiwan? The predominant (∼77%) chicken mtDNA lineage in the Pacific—haplogroup D—is absent in Taiwan (Figure 3; Chang et al., 2012). Thus, the Pacific lineages belonging to haplogroup D are most likely to originate from Southeast Asia. In support of this, a substantial introduction of domesticates from Southeast Asia into Pacific is revealed by analyzing mtDNA data of the dog (Oskarsson et al., 2012), pig (Lum et al., 2006; Larson et al., 2007, 2010) and even the commensal rat (Matisoo-Smith and Robins, 2004). Consequently, Southeast Asia likely served as the homeland of most domesticates spreading to the Pacific. The recruitment of multiple local domesticates at least indicates the substantial cultural contributions from Southeast Asia.
In summary, the chicken mtDNA phylogeny in the present study provides the highest level of molecular resolution to date. The reconstructed matrilineal phylogeny covers most of the major mtDNA haplogroups in chickens. It serves as a basis for (re)assessing the haplogroup variation of modern breeds, as well as well-preserved ancient remains. Our results expand the perspectives into the complex history of chicken domestication and dissemination. Future zooarchaeological investigations and multidisciplinary research (Zeder et al., 2006; Matisoo-Smith, 2009), including comprehensive genetic studies, are required to unravel more details.
Data archiving
All mtDNA control region and complete genome sequences were deposited in GenBank with accession numbers GU261674–GU261719 and GU447321–GU449100.
Accession codes
References
Achilli A, Bonfiglio S, Olivieri A, Malusa A, Pala M, Kashani BH et al (2009). The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS One 4: e5753.
Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N et al (2008). Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr Biol 18: R157–R158.
Achilli A, Olivieri A, Soares P, Lancioni H, Hooshiar KB, Perego UA et al (2012). Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc Natl Acad Sci USA 109: 2449–2454.
Behar DM, van Oven M, Rosset S, Metspalu M, Loogväli EL, Silva NM et al (2012). A ‘copernican’ reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet 90: 675–684.
Bellwood P (1976). Prehistoric plant and animal domestication in Austronesia. In: GdG Sieveking, Longworth IH, Wilson ED, (eds) Problems in Economic and Social Archaeology. Westview Press Boulder, CO. pp 153–168.
Berthouly C, Leroy G, Van TN, Thanh HH, Bed'Hom B, Nguyen BT et al (2009). Genetic analysis of local Vietnamese chickens provides evidence of gene flow from wild to domestic populations. BMC Genet 10: 1.
Bonfiglio S, Achilli A, Olivieri A, Negrini R, Colli L, Liotta L et al (2010). The enigmatic origin of bovine mtDNA haplogroup R: sporadic interbreeding or an independent event of Bos primigenius domestication in Italy? PLoS One 5: e15760.
Chang CS, Chen CF, Berthouly-Salazar C, Chazara O, Lee YP, Chang CM et al (2012). A global analysis of molecular markers and phenotypic traits in local chicken breeds in Taiwan. Anim Genet 43: 172–182.
Chang H (2009) Animal Genetic Resources. Science Press Beijing.
Crawford RD (1990). Origin and history of poultry species. In: Crawford RD, (ed) Poulty Breeding and Genetics. Elsevier Amsterdam. pp 1–59.
Dancause KN, Vilar MG, Steffy R, Lum JK (2011). Characterizing genetic diversity of contemporary pacific chickens using mitochondrial DNA analyses. PLoS One 6: e16843.
Darwin C (1896). The Variation of Animals and Plants under Domestication. 2nd edn. Vol 1., D. Appleton: New York.
Desjardins P, Morais R (1990). Sequence and gene organization of the chicken mitochondrial genome. A novel gene order in higher vertebrates. J Mol Biol 212: 599–634.
Diamond JM (2000). Taiwan's gift to the world. Nature 403: 709–710.
Eriksson J, Larson G, Gunnarsson U, Bed’hom B, Tixier-Boichard M, Stromstedt L et al (2008). Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet 4: e1000010.
FAO (2007). The State of the World’s Animal Genetic Resources for Food and Agriculture,. FAO Rome.
Forster P, Harding R, Torroni A, Bandelt HJ (1996). Origin and evolution of native American mtDNA variation: a reappraisal. Am J Hum Genet 59: 935–945.
Fumihito A, Miyake T, Sumi S, Takada M, Ohno S, Kondo N (1994). One subspecies of the red junglefowl (Gallus gallus gallus) suffices as the matriarchic ancestor of all domestic breeds. Proc Natl Acad Sci USA 91: 12505–12509.
Fumihito A, Miyake T, Takada M, Shingu R, Endo T, Gojobori T et al (1996). Monophyletic origin and unique dispersal patterns of domestic fowls. Proc Natl Acad Sci USA 93: 6792–6795.
Hurles ME, Matisoo-Smith E, Gray RD, Penny D (2003). Untangling Oceanic settlement: the edge of the knowable. Trends Ecol Evol 18: 531–540.
Hutagalung RI (2000). The monetary crisis and its impact on the development of the poultry industry in Indonesia. Proc Aust Poult Sci Syn 12: 74–81.
Kanginakudru S, Metta M, Jakati RD, Nagaraju J (2008). Genetic evidence from Indian red jungle fowl corroborates multiple domestication of modern day chicken. BMC Evol Biol 8: 174.
Kivisild T, Shen P, Wall DP, Do B, Sung R, Davis K et al (2006). The role of selection in the evolution of human mitochondrial genomes. Genetics 172: 373–387.
Komiyama T, Ikeo K, Gojobori T (2003). Where is the origin of the Japanese gamecocks? Gene 317: 195–202.
Larson G, Cucchi T, Fujita M, Matisoo-Smith E, Robins J, Anderson A et al (2007). Phylogeny and ancient DNA of Sus provides insights into neolithic expansion in Island Southeast Asia and Oceania. Proc Natl Acad Sci USA 104: 4834–4839.
Larson G, Liu R, Zhao X, Yuan J, Fuller D, Barton L et al (2010). Patterns of East Asian pig domestication, migration, and turnover revealed by modern and ancient DNA. Proc Natl Acad Sci USA 107: 7686–7691.
Liu YP, Wu GS, Yao YG, Miao YW, Luikart G, Baig M et al (2006). Multiple maternal origins of chickens: out of the Asian jungles. Mol Phylogenet Evol 38: 12–19.
Lum JK, McIntyre JK, Greger DL, Huffman KW, Vilar MG (2006). Recent Southeast Asian domestication and Lapita dispersal of sacred male pseudohermaphroditic ‘tuskers’ and hairless pigs of Vanuatu. Proc Natl Acad Sci USA 103: 17190–17195.
Macdonald KC, Blench RM (2000). Chickens. In: Kiple KF, Ornelas KC, (eds). The Cambridge Word History of Food Vol. 1, Cambridge University Press Cambridge. pp 496–499.
Matisoo-Smith E (2009). The commensal model for human settlement of the Pacific 10 years on—what can we say and where to now? J Island Coastal Arch 4: 151–163.
Matisoo-Smith E, Robins JH (2004). Origins and dispersals of Pacific peoples: evidence from mtDNA phylogenies of the Pacific rat. Proc Natl Acad Sci USA 101: 9167–9172.
Nishibori M, Shimogiri T, Hayashi T, Yasue H (2005). Molecular evidence for hybridization of species in the genus Gallus except for Gallus varius. Anim Genet 36: 367–375.
Oka T, Ino Y, Nomura K, Kawashima S, Kuwayama T, Hanada H et al (2007). Analysis of mtDNA sequences shows Japanese native chickens have multiple origins. Anim Genet 38: 287–293.
Oskarsson MC, Klutsch CF, Boonyaprakob U, Wilton A, Tanabe Y, Savolainen P (2012). Mitochondrial DNA data indicate an introduction through Mainland Southeast Asia for Australian dingoes and Polynesian domestic dogs. Proc Biol Sci B 279: 967–974.
Pang JF, Kluetsch C, Zou XJ, Zhang AB, Luo LY, Angleby H et al (2009). mtDNA data indicate a single origin for dogs south of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol Biol Evol 26: 2849–2864.
Patrick VK (2010). Peopling of the Pacific: a holistic anthropological perspective. Annu Rev Anthropol 39: 131–148.
Pereira L, Freitas F, Fernandes V, Pereira JB, Costa MD, Costa S et al (2009). The diversity present in 5140 human mitochondrial genomes. Am J Hum Genet 84: 628–640.
Saillard J, Forster P, Lynnerup N, Bandelt HJ, Norby S (2000). mtDNA variation among Greenland Eskimos: the edge of the Beringian expansion. Am J Hum Genet 67: 718–726.
Soares P, Ermini L, Thomson N, Mormina M, Rito T, Röhl A et al (2009). Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet 84: 740–759.
Storey AA, Ladefoged T, Matisoo-Smith EA (2008). Counting your chickens: density and distribution of chicken remains in archaeological sites of Oceania. Int J Osteoarchaeol 18: 240–261.
Tamura K, Dudley J, Nei M, Kumar S (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
van Oven M, Kayser M (2009). Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30: E386–E394.
West B, Zhou B-X (1988). Did chickens go North? New evidence for domestication. J Arch Sci 15: 515–533.
Wu GS, Yao YG, Qu KX, Ding ZL, Li H, Palanichamy MG et al (2007). Population phylogenomic analysis of mitochondrial DNA in wild boars and domestic pigs revealed multiple domestication events in East Asia. Genome Biol 8: R245.
Wu Z, Kaiser P (2011). Antigen presenting cells in a non-mammalian model system, the chicken. Immunobiology 216: 1177–1183.
Yuan J (2010). Zooarchaeological study on the domestic animals in ancient China. Quaternary Sci 30: 298–306.
Zeder MA, Emshwiller E, Smith BD, Bradley DG (2006). Documenting domestication: the intersection of genetics and archaeology. Trends Genet 22: 139–155.
Zeuner FE (1963) A History of Domesticated Animals. Harper & Row New York.
Acknowledgements
We are grateful to volunteers for collecting samples. We thank Shi-Fang Wu, Wen-Zhi Wang and Kai He for technical assistance. This study was supported by grants from the National Basic Research Program of China (973 Program, 2007CB815704), the National Natural Science Foundation of China (U0836602, 31061160189 and 30660024) and the Natural Science Foundation of Yunnan Province, China (2007C0003Z and 2006C0034M). This work was also supported, in part, by a Visiting Professorship for Senior International Scientists from the Chinese Academy of Sciences and manuscript preparation by a Natural Sciences and Engineering Research Council of Canada Discovery Grant A3148 to RWM. MB is thankful to CAS-TWAS for postdoctoral fellowship award 2007 to work at Kunming Institute of Zoology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Miao, YW., Peng, MS., Wu, GS. et al. Chicken domestication: an updated perspective based on mitochondrial genomes. Heredity 110, 277–282 (2013). https://doi.org/10.1038/hdy.2012.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.83
Keywords
This article is cited by
-
High quality assemblies of four indigenous chicken genomes and related functional data resources
Scientific Data (2024)
-
Origin and spatial population structure of Malagasy native chickens based on mitochondrial DNA
Scientific Reports (2024)
-
Archaeological and molecular evidence for ancient chickens in Central Asia
Nature Communications (2024)
-
VDR promotes testosterone synthesis in mouse Leydig cells via regulation of cholesterol side chain cleavage cytochrome P450 (Cyp11a1) expression
Genes & Genomics (2023)
-
Introducing the land snail Bradybaena pellucida increased infection risk of the avian parasite Postharmostomum commutatum in the Kanto region of Japan
Parasitology Research (2023)
