
Abstract
House sparrow (Passer domesticus) populations have suffered major declines in urban as well as rural areas, while remaining relatively stable in suburban ones. Yet, to date no exhaustive attempt has been made to examine how, and to what extent, spatial variation in population demography is reflected in genetic population structuring along contemporary urbanization gradients. Here we use putatively neutral microsatellite loci to study if and how genetic variation can be partitioned in a hierarchical way among different urbanization classes. Principal coordinate analyses did not support the hypothesis that urban/suburban and rural populations comprise two distinct genetic clusters. Comparison of FST values at different hierarchical scales revealed drift as an important force of population differentiation. Redundancy analyses revealed that genetic structure was strongly affected by both spatial variation and level of urbanization. The results shown here can be used as baseline information for future genetic monitoring programmes and provide additional insights into contemporary house sparrow dynamics along urbanization gradients.
Similar content being viewed by others

Introduction
Population connectivity, mediated by dispersal, affects various ecological and evolutionary key features, such as population growth rates, spatial distribution of genetic diversity, local adaptation and global dynamics (Ronce, 2007). Because population connectivity shapes the resilience of populations against demographic, genetic and environmental disturbance (Hanski, 1999), it also constitutes a key determinant of their long-term viability. Assessing dispersal rates using capture–mark–recapture is often cumbersome and time-consuming, and low probabilities of resighting or retrapping due to the inaccessibility of open areas (for example, private gardens) and strong human disturbance in urban centers, may additionally hamper its use. Nowadays, the wealth of genetic markers, which are readily available for most species and the advent of new statistical analytical methods have provided researchers with a powerful alternative to indirectly measure dispersal using genotype data (Neigel, 1997; Prugnolle and de Meeus, 2002). In addition, a body of literature has presented numerous correlations between genetic variation and fitness traits, such as growth, survival and disease resistance (Falconer and MacKay, 1996; Reed and Frankham, 2003), and many conservation biologists are therefore concerned in preserving the remnant genetic variation within a species. Knowledge on the geographical distribution of existing genetic variation has thus become an invaluable source of information when aiming to delineate appropriate biological conservation units (Taylor and Dizon, 1996; Gebremedhin et al., 2009). A solid understanding of genetic population substructure and exchange of migrants between populations may complement demographic studies and has become a major goal of many ecological studies as it can inform both population ecologists and conservation managers.
One of the species that has recently drawn the attention of conservation biologists is the house sparrow (Passer domesticus). Once a thriving ubiquitous species (Anderson, 2006), house sparrows have suffered a dramatic decline in abundance and distribution during the last decades (Hole et al., 2002; Chamberlain et al., 2007; De Laet and Summers-Smith, 2007) and evidence has mounted that these reductions vary considerably in space and time (De Laet and Summers-Smith, 2007; Shaw et al., 2008). Within strongly built-up areas, urban populations, which have suffered massive declines, can generally be differentiated from suburban ones, which have remained rather stable. Apart from the extent of the population decline, the onset in rural areas also preceded that in the urban one, and although rural population numbers seem to have stabilized (albeit at lower population equilibria), urban populations continue to plummet (De Laet and Summers-Smith, 2007). Yet, despite this demographic heterogeneity, post-decline data on small-scale genetic structuring along urbanization gradients are still lacking for this species. This paucity of information may hamper future conservation efforts and advocates for an urgent need to bridge this lacuna.
House sparrows are among the most sedentary of all passerine birds, being characterized by short postnatal dispersal distance (1–1.7 km; Anderson, 2006) and strong levels of site tenacity after their first breeding attempt (Summers-Smith, 1988). Based on this sedentary behavior and the difference in timing, magnitude and progress of the population decline, urban/suburban and rural populations are thought to comprise two distinct and independent units that may call for different conservation strategies (Anderson, 2006; De Laet and Summers-Smith, 2007; Shaw et al., 2008). Such dichotomy implicitly assumes that both habitats are characterized by a high degree of isolation and are to a large extent self-sustainable (Wilson, 2004). Likewise, the small-scale heterogeneity in demographic features within the built-up environment at least casts doubt that urban and suburban areas constitute a single independent unit. This view, however, was challenged by Heij (1985) who concluded that both urban and rural study populations were highly dependent on suburban immigrants for their long-term viability. Several other studies also failed to reveal (large) genetic dissimilarities between house sparrow populations in the absence of strong geographical barriers (Fleisher, 1983; Parkin and Cole, 1984; Kekkonen et al., 2010). Yet, all these studies had in common that they described dispersal and/or genetic structure well before the onset of the population decline.
In contrast, Hole (2001) found low but significant genetic structuring and loss of connectivity between neighboring farms in a rural area after the decline had commenced. Local extinction without recolonization may transform contiguous populations into patchy ones, a pattern currently (mainly) observed in urban areas (Shaw et al., 2008; Vangestel, 2011). As house sparrows most likely (used to) disperse according to a stepwise pattern (Kekkonen et al., 2010), loss of intermediate ‘stepping-stones’ combined with the intrinsic sedentary nature of house sparrows (Anderson, 2006) may have reduced or even inhibited contemporary dispersal between adjacent colonies (Hole, 2001). Hole (2001) was among the first to suggest that such loss of (intermediate) populations may gradually increase distances between remaining source–sink populations up to a point at which house sparrow metapopulation dynamics become constrained and populations genetically diverge, ultimately resulting in an extinction vortex that spreads through the landscape. Less vagile species (such as the house sparrow), in particular, are expected to be highly vulnerable to such landscape alterations (Hole, 2001; Sekercioglu et al., 2002) and to show a tendency towards strong local population structuring (Stangel, 1990). Given the observed large-scale reductions in urban house sparrow numbers (De Laet and Summers-Smith, 2007), we could hence expect that contemporary urban house sparrow populations are particularly susceptible to genetic drift, while exchange of migrants between contemporary populations may become problematic.
Here we quantify genetic variation across microsatellite loci to test whether house sparrows show small-scale genetic differentiation and changes in genetic diversity along an urban-rural gradient, and to relate this to putative demographic changes. Besides its relevance for future conservation strategies, these results may further contribute to our general understanding of population dynamics of species in contemporary anthropogenic landscapes.
Materials and methods
Study site and species
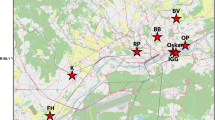
House sparrows were sampled in the greater area of Ghent (northern Belgium) and in an adjacent rural area near the village of Zomergem, ca. 12 km northwest of Ghent. The degree of urbanization, measured as the ratio of built-up to total grid cell area (each cell measuring 90 000 m2 on the ground), ranged between 0–0.10 (henceforth referred to as ‘rural’ area), 0.11–0.30 (‘suburban’ area) and >0.30 (‘urban’ area), respectively (Arcgis version 9.2, ESRI, Redlands, CA, USA) (Figure 1). Although we currently lack long-term data on house sparrow densities within our study area, we believe it is reasonable to expect substantial demographic heterogeneity along the urbanization gradient for the following reasons. First, earlier studies (Vangestel et al., 2010; Vangestel, 2011) within the same study area suggested that house sparrows experience the urban environment as the most stressful one, based on the slower feather growth rates measured in urban compared with suburban and rural populations. This phenotypic trait was earlier shown to be positively related to a range of fitness components and to comprise a reliable proxy for nutritional condition and energetic stress (Grubb, 2006 and references therein). Secondly, census counts using a mapping-based survey technique revealed equally low urban house sparrow density (0.57 birds ha−1) as in other large cities, such as London and Hamburg, which have well-documented house sparrow declines. In contrast, suburban and rural population sizes were several magnitudes larger (Vangestel, 2011). Between the years 2003 and 2009, a total of 690 adult house sparrows were captured by standard mist netting in 26 study plots in rural, suburban and urban areas (roughly equal sex ratios obtained per plot). As there was no evidence that allele frequencies substantially shifted during the course of the sampling period, samples were pooled across years. Upon capture, standard morphological measurements were taken (details in Vangestel et al., 2010) and a small sample of body feathers was collected for DNA analysis.

Map showing the location of the 26 urban (filled circles), suburban (open circles) and rural (filled triangles) populations under study. The inner contour encompasses the city center of Ghent, the outer contour encompasses the surrounding municipalities.
Morphological variation
A general linear mixed model was applied to study variation in average body weight between urbanization classes. Sex, month and hour were added as fixed covariates, whereas year and study plot were modeled as random factors to account for a possible clustered data structure. Measures for body condition were obtained by adding tarsus size as a fixed covariate to the previous model (Jakob et al., 1996). All statistical analyses were performed in SAS v9.2 (SAS Institute 2008, Cary, NC, USA).
DNA extraction, PCR and genotyping
We applied a Chelex resin-based method (InstaGene Matrix, Bio-Rad, Hercules, CA, USA) (Walsh et al., 1991) to extract genomic DNA from a total of ten plucked body feathers per individual. In all, 16 microsatellite markers (both traditional ‘anonymous’ microsatellites as well as those developed based on expressed sequence tags; Table 1) were selected based on their polymorphism and stutter profile. Polymerase chain reactions were organized in four multiplex sets and compatibility between primer pairs was checked using AutoDimer (Vallone and Butler, 2004). The first multiplex reaction contained Pdoμ1 (Neumann and Wetton, 1996), Pdo32, Pdo47 (Dawson et al., 2012) and TG04-012 (Dawson et al., 2010); the second one contained Pdoμ3 (Neumann and Wetton, 1996), Pdoμ5 (Griffith et al., 1999), TG13-017 and TG07-022 (Dawson et al., 2010); the third multiplex reaction contained Pdo10 (Griffith et al., 2007), Pdo16, Pdo19, Pdo22 (Dawson et al., 2012) and TG01-040 (Dawson et al., 2010); the last set consisted of Pdo9 (Griffith et al., 2007), TG01-148 and TG22-001 (Dawson et al., 2010). PCR reactions were performed on a 2720 Thermal Cycler (Applied Biosystems, Foster City, CA, USA) in 9 μl volumes and contained approximately 3 μl (30 ng) of genomic DNA, 3 μl QIAGEN Multiplex PCR Mastermix (Qiagen, Venlo, The Netherlands) and 3 μl primer mix (concentrations were 0.1 μM (Pdoμ1), 0.12 μM (TG01-148), 0.16 μM (Pdo10, Pdo19, Pdo22, Pdo32, TG04-012) and 0.2 μM (Pdoμ3, Pdoμ5, Pdo9, Pdo16, Pdo47, TG01-040, TG07-022, TG13-017, TG22-001). The PCR profile contained an initial denaturation step of 15 min at 95 °C, followed by 35 cycles of 30 s at 94 °C, 90 s at 57 °C and 60 s at 72 °C. Finally, an additional elongation step of 30 min at 60 °C and an indefinite hold at 4 °C was included. Prior to genotyping, the DNA concentration of each sample was quantified using a ND1000 spectrometer (Nanodrop Technologies, Wilmington, DE, USA) and adjusted to a standard concentration of 10 ng μl−1. To rule out contamination of reagents, negative controls were applied during extraction and PCR. PCR products were visualized on an ABI3730 Genetic Analyzer (Applied Biosystems), an internal LIZ-600 size standard was applied to determine allele size, standard samples were included in each run and fragments were scored using the software package GENEMAPPER 4.0 (Applied Biosystems). All loci under study were autosomal as inferred from the chromosomal location of their homologs on the genome of the zebra finch, Taeniopygia guttata (Vangestel et al. unpublished data) and confirmed autosomal in house sparrow by the successful amplification of the loci in both sexes and by the presence of heterozygotes in female individuals for each locus.
We used MICRO-CHECKER (Van Oosterhout et al., 2004) to identify scoring errors that could be attributed to stuttering, differential amplification of size-variant alleles causing large allele drop-out or the presence of null alleles. All microsatellite loci were checked for Hardy–Weinberg and linkage equilibrium with GENEPOP 4.0 (Raymond and Rousset, 1995; Rousset, 2008) and a family-wise error rate of 0.05 was obtained by applying a Bonferroni correction (Weir, 1990).
Genetic diversity and population structure
For each locus-by-population combination, genetic diversity was quantified by allelic richness (A[g]), observed heterozygosity (Ho) and unbiased expected heterozygosity (He) (Nei, 1978). Measures of heterozygosity were computed in FSTAT 2.9.3.2 (Goudet, 1995), whereas allelic richness was calculated with ARES 1.2.2 (Van Loon et al., 2007). As estimates of allelic richness vary with sample size (Kalinowski, 2004) and patterns can be obscured or even reversed when samples of different sizes are compared (Van Loon et al., 2007), a rarefaction procedure was applied (sensu Hurlbert, 1971). Estimates were extrapolated to the average number of gene copies per population (g=40), rather than to the smallest observed sample size, which might result in a loss of information or decrease in accuracy (Van Loon et al., 2007). Heterogeneity in genetic diversity between urban, suburban and rural populations was tested by running 1000 permutations. We calculated FST indices (Weir and Cockerham, 1984) using Genetix 4.05.2 (Belkhir et al., 2004) to assess pairwise genetic differentiation among all populations and overall genetic differentiation within each urbanization class. The significance of pairwise FST values was evaluated using 1000 permutations.
We conducted an individual-based Bayesian analysis implemented with STRUCTURE 2.2 (Pritchard et al., 2000) to delineate clusters (K) of individuals based on their multilocus genotypes. Without a priori knowledge of source populations, this algorithm groups individuals by minimizing deviation from Hardy–Weinberg and linkage disequilibrium (Pritchard et al., 2000). We applied an admixture model with ten independent runs of K=1–26, 200 000 Markov Chain Monte Carlo repetitions, a burn-in period of 100 000, correlated allele frequencies and no prior information on the population of origin. The modal value of the ad hoc quantity ΔK, based on the second order rate of change of the likelihood function (ln Pr (X|K)), was used as criterion to detect the true K (Evanno et al., 2005). This approach overcomes difficulties in estimating the true K in case likelihood values increase with stepwise values of K (Pritchard et al., 2007). When ΔK failed to reveal an unambiguous signal, we selected the model with the highest mean value of ln Pr (X|K) as the most parsimonious one (Pritchard et al., 2007).
Next, we applied an alternative clustering algorithm with INSTRUCT (Gao et al., 2007). This extension of STRUCTURE does not assume Hardy–Weinberg equilibrium within clusters and jointly estimates levels of population inbreeding and population structure. Apart from estimating a population inbreeding coefficient, the settings used were identical to those applied in STRUCTURE. Replicate cluster analyses were aligned with CLUMPP 1.1.2 (Jakobsson and Rosenberg, 2007) and visualized with DISTRUCT 1.0 (Rosenberg, 2004). Subsequently, we conducted a Principal Coordinate Analysis (PCoA) to summarize pairwise FST values obtained from an Analysis of Molecular Variance (AMOVA) as implemented in GENALEX 6.4 (Peakall and Smouse, 2006). This multivariate dimension-reducing method is exploratory in nature and graphical displays of the most important principal axes were used to visualize genetic similarity among populations within urbanization classes (Jombart et al., 2009). Finally, we conducted an AMOVA in ARLEQUIN 3.5.1.2 (Excoffier et al., 2005) to examine how genetic variation was partitioned over three hierarchical scales: within populations, between populations within urbanization classes and among urbanization classes. As the statistical power of a nested AMOVA depends on the number of populations that can be permutated among higher hierarchical levels (rather than on the number of individuals or loci), we first applied the multinomial theorem to our sampling scheme to ascertain that the minimum P-value was adequately small (Fitzpatrick, 2009).
A hierarchical analysis of genetic variance across spatial scales provides a first estimate of a deviation from migration-drift equilibrium, and of the extent of dispersal limitation. Here, three spatial clusters are present: rural, suburban and urban samples. If a metapopulation is at or near migration-drift equilibrium, genetic differentiation at a lower hierarchical level (within clusters) cannot exceed the genetic differentiation at a higher hierarchical level (among clusters). If on the other hand, genetic differentiation among populations within a spatial cluster is larger than among clusters, this provides proof that the genetic structure is more determined by genetic drift than by dispersal. This follows from the principle that drift is stronger in small populations (local scale) than in large populations (cluster scale). Hence, such patterns represent a deviation from migration-drift equilibrium, reflecting a future loss of genetic diversity (genetic extinction debt).
To investigate in more detail the extent to which the genetic structure (genetic distance) and genetic diversity was affected by spatial structure (dispersal limitation) on the one hand, and the degree of urbanization on the other hand, we performed a redundancy analysis (RDA) in Canoco 4.5 (ter Braak and Šmilauer, 2002). As dependent variables, we used genetic structure (PCoA loadings of the first six PCoA axes for each population), and in a separate analysis measures of genetic variation (He per locus). As explanatory variables, we used two sets of variables. The first is the spatial variable set (S), consisting of the geographic locations of the populations in a two-dimensional grid (x, y) and transformations thereof (Borcard et al., 1992): xy, x2, y2, x2y, xy2, x3, y3, ln(x) and ln(y). The second set represents the urbanization degree (U) in a radius of 50, 250 and 1250 m around the population centers (hence labeled U50, U250 and U1250, respectively). Using these two sets of explanatory variables, we performed variance partitioning (Borcard et al., 1992) to decompose the total explained variance (in genetic structure and genetic diversity, respectively) into the unique contribution of spatial variables (S|U), of urbanization variables (U|S) and the intersection of both (S∩U). We used an automatic forward selection procedure implemented in Canoco to maximally improve the fit of each model. In addition, we estimated the unique contribution of each variable to the model and the significance thereof using 999 monte carlo permutations. Finally, we determined the unique contribution of spatial variables and of urbanization variables in reduced models, using only variables that contributed significantly to the earlier full models. This approach is much more powerful than (partial) Mantel tests as these use only a fraction of the available information (all variation is compressed into a single variable), whereas multivariate ordination approaches such as RDA use more information and are much more sensitive to subtle patterns (Fortin and Dale, 2005).
Population bottlenecks and dispersal
To detect genetic signatures of recent population bottlenecks, we visually inspected allele frequency distributions for mode shifts from low to intermediate allele frequency with BOTTLENECK 1.2.02 (Cornuet and Luikart, 1996; Piry et al., 1999). Strong reductions in effective population size result in contrasting rates at which allelic diversity and heterozygosity at Hardy–Weinberg equilibrium (He) are lost (Cornuet and Luikart, 1996), with particularly strong losses of rare alleles. Whereas the latter has a small impact on He only (Hedrick et al., 1986), it strongly affects the expected heterozygosity at mutation-drift equilibrium (Heq) as these estimates depend on the absolute number of alleles (Ewens, 1972), and the transient disruption of mutation-drift equilibrium in bottlenecked populations generates an excess in heterozygosity (He>Heq) (Luikart and Cornuet, 1998). As Heq distributions obtained through coalescence-based simulations strongly hinge on the (unknown) model of microsatellite evolution (Hawley et al., 2006), the occurrence of a bottleneck was assessed by visual inspection (mode shift) rather than formal testing.
To quantify recent gene flow between urban, suburban and rural areas, we pooled all populations within a particular urbanization class into a single population cluster and identified the cluster of origin for each individual by computing posterior probability densities of unknown population allele frequencies and subsequent individual multilocus genotype probabilities (partial assignment technique sensu Rannala and Mountain, 1997). Next, we generated a likelihood distribution of simulated genotypes and excluded a particular population cluster as possible origin if the likelihood of a genotype fell in the tail-end of this distribution (Cornuet et al., 1999). By excluding two of three clusters the putative source of an individual could be identified, and this individual was considered a ‘disperser’ if the population in which it was captured differed from the presumed cluster of origin (Cornuet et al., 1999). Although the statistical power of assignment tests ultimately depends on the level of population differentiation (Paetkau et al., 2004), these tests are believed to be robust against deviations from migration-drift equilibrium as assignment is performed at an ecological (not evolutionary) timescale (Peery et al., 2008). Analyses were performed in GENECLASS 2.0 (Piry et al., 2004) and focal individuals were removed from the sample when allele frequencies were estimated (‘leave-one-out’ procedure).
Results
Neither weight nor body condition varied significantly between urbanization classes (respectively F2,17.1=0.65; P=0.54 and F2,17.4=0.66; P=0.53). All locus-by-population combination were highly polymorphic, with numbers of alleles per locus ranging between 3 (TG01-148) and 31 (Pdo9). Apart from Pdo47 (three populations), Pdoμ5 (two populations) and Pdo9, Pdo32, TG01-148, TG07-022 and TG13-017 (one population each), all locus-by-population combinations were in Hardy–Weinberg equilibrium after Bonferroni correction. There was no evidence that disequilibria in the loci mentioned above reflected scoring errors due to large allele drop-out or stutter, and removing them did not alter any of the observed patterns, hence results based on the analysis of all loci are presented. There was also no evidence for linkage disequilibrium between any pair of loci (Table 1).
Genetic diversity and population structure
Mean numbers of alleles per locus ranged between 4.8 and 7.9, allelic richness ranged between 5.86 and 9.62 and expected heterozygosity ranged between 0.60 and 0.71. Based on permutation tests, none of these indices differed significantly between urbanization classes (all P>0.05). Levels of pairwise population differentiation ranged between −0.028 and 0.120 (mean FST±s.e.m.: 0.018±0.001) (Table 2). When pooling all populations per urbanization class, pairwise values of the differentiation index FST equaled 0.0019 (urban-rural), 0.0013 (suburban-rural) and 0.0007 (urban-suburban), whereas overall levels of differentiation within each cluster equaled 0.0138 (urban), 0.0119 (suburban) and 0.0267 (rural), respectively. Thus, genetic differentiation within each cluster is an order of magnitude higher than among clusters, which is evidence for strong deviation from migration-drift equilibrium.
Details of all RDA analyses are presented in the Supplementary Material (Supplementary Appendix S1). Redundancy analyses on genetic diversity with both spatial and urbanization variables explained 36.2% of the variance in genetic diversity found in populations (expected heterozygosity per locus), although no single variable contributed significantly to the model (P>0.07). The variance partitioning showed that spatial variables alone explained 6.7%, though no single variable had a significant contribution. Urbanization uniquely explained 11.5% of the variance, but again no variable contributed significantly.
When considering genetic structure based on the PCoA loadings of each population, spatial and urbanization variables jointly explained 34.6% of the variance in genetic structure. Two urbanization and one spatial variable contributed significantly, yielding a reduced model that explained 31.4% of the total variance (P=0.001). Given that urbanization variables were included in the model, spatial variables uniquely explained 34.0% of the total variance, with a single significant variable (y2) explaining 20% of the variance in genetic structure (P=0.001). The unique contribution of urbanization (U|S) was 18.4%, of which U250 was retained in a reduced model (R2=11.4%, P=0.02).
When the RDA on the genetic structure was limited to rural populations or (sub)urban populations, a (marginally) significant spatial pattern emerged after forward selection of the variable y3 (rural R2=0.19, P=0.036) or the variable y ((sub)urban R2=0.17, P=0.046). When this was repeated for the degree of urbanization per cluster while correcting for spatial covariation, patterns persisted in (sub)urban areas (variable U250, R2=0.23, P=0.03), but not in the rural one.
We failed to identify distinct population clusters along the urbanization gradient, which is consistent with the low level of genetic differentiation among populations. Whereas clustering in STRUCTURE did not show a clear mode for ΔK, posterior probabilities gradually declined from K>7 onwards, with K=1 selected as the most parsimonious model (Figures 2a–d). When taking variation in inbreeding into account, posterior probabilities incremented with increasing values of K and reached a plateau near K=13. To overcome this problem, we used ΔK to delineate the most appropriate model. This, however, did not reveal a clear mode either, although lower numbers of clusters were associated with the largest ΔK values (K=1 and K=2, respectively) (Figures 2e–h).

Graphical display of the four steps to detect the true number of clusters (following Evanno et al., 2005). (a) and (e): L(K), mean posterior likelihood (±s.d.) over 10 runs for each K value. (b) and (f): L′(K), first order rate of change in the posterior likelihood (±s.d.). (c) and (g): L′′(K), absolute value of the second order rate of change in the posterior likelihood (±s.d.). (d) and (h): ΔK, mean absolute values of L′′(K) over s.d. of L(K). (a–d): STRUCTURE analysis (Pritchard et al., 2000, e–h): INSTRUCT analysis (Gao et al., 2007).
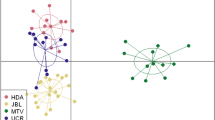
Lack of support for strong genetic population structuring was confirmed by the fact that very few individuals showed a unique affinity to a specific urbanization class. Overall, PCoA analysis revealed weak clustering of individuals from adjacent populations but did not support strong clustering of the different urbanization classes. Both main PCoA axes together explained 83.74% of the total genetic variation (Figure 3). Based on a hierarchical AMOVA model, 0% genetic variation was assigned to differences between urbanization classes, 2.2% variation to between population variation within urbanization classes and 97.8% variation to variation within populations. As the minimum P-value for three groups of 13, 9 and 4 populations was much smaller than 0.001, lack of support for genetic population structure at higher hierarchical levels was not due to low statistical power.

Scatter plot of the first two PCoA axes of genetic variation (based on pairwise FST values) between 26 house sparrow populations. Filled circles (urban populations), open circles (suburban populations), filled triangles (rural populations).
Population bottlenecks and gene flow
Of the 26 populations assessed, only the most central urban population (EB-U) showed genetic evidence of a recent population bottleneck. Here, allelic richness was low whereas heterozygosity was retained at a level equivalent to that of other populations, and a clear mode was present at intermediate (rather than low) frequency alleles. In all other populations, allele frequency distributions were typically L-shaped (see Supplementary Appendix S2).
Because of the weak level of genetic population structuring, statistical power to quantify gene flow among urbanization classes was low as well. Only 10.1% of all individuals (rural 31 individuals; suburban 32 individuals; urban 7 individuals) were unambiguously assigned to a single source cluster. Among the rural individuals, 61% were assigned as resident, whereas 39% were assigned as immigrant from the suburban population cluster. Among the suburban individuals, 78% were assigned as resident and 22% were assigned as immigrant from the rural cluster. Finally, among the urban individuals, all individuals were assigned as immigrant, 43% of which originated from the suburban cluster and 57% from the rural cluster. Most of the other individuals showed a putative mixed ancestry (73.9%), whereas a smaller fraction (15.9%) was assigned as immigrant from an unknown population source (Table 3).
Discussion
Despite the rapid and steep decline of house sparrow populations in many metropolitan areas across Europe (De Laet and Summers-Smith, 2007 and references therein), genetic population studies remain remarkably scant. Spatial analysis of putatively neutral microsatellite genotypes in 26 house sparrow populations along an urbanization gradient in Flanders revealed evidence for high historical connectivity or recent common ancestry. Redundancy analyses showed that local genetic diversity per sé was not significantly affected by dispersal limitation (spatial variables) or by the degree of urbanization, but the genetic structure, that is, how the regional genetic diversity was distributed across the landscape, was significantly affected by both spatial variation (dispersal limitation) and the degree of urbanization. The most central urban population showed a genetic signature of a recent bottleneck, however, no such signal was apparent in more peripheral urban populations. Small census sizes in the urban periphery (Vangestel, 2011) and the suggested unidirectional gene flow from suburban and rural populations into urban peripheral ones, further support the hypothesis that urban populations act as demographic sinks in which genetic drift is (at least partly) countered by gene flow.
The spatially hierarchical analysis of genetic differentiation, with much higher differentiation among nearby populations than among more distant clusters, showed evidence for strong deviations from migration-drift equilibrium. This indicates that genetic drift is currently a much more important process than gene flow. Consequently, we can expect a future reduction in genetic diversity, especially among clusters. Contrary to our expectation, we found no evidence of stronger isolation and decrease in numbers in urban populations compared with suburban or rural ones. This may either indicate that urban populations remained stable in the recent past—possibly at a lower carrying capacity—or that we failed to pick up the genetic signal associated with a demographic population bottleneck. However, the bottleneck test assumes a closed population, whereas this is likely not the case, compromising our potential to detect demographic bottleneck events (Keller et al., 2001). Unfortunately, lack of historical data series prevents us from testing this hypothesis directly. Dispersal rates estimated from individual-based assignment tests indicated moderate site tenacity in suburban and rural populations, supporting the presumed sedentary status of house sparrows (Anderson, 2006). A complete lack of immigrants from urban populations in either of these populations, however, was unexpected, especially in view of the fact that the urban population cluster consisted exclusively of immigrants from suburban and rural populations. Such unidirectional gene flow provides additional evidence for an urban ‘dispersal sink’ (sensu Dias, 1996) that may have masked bottleneck signatures in all urban populations apart from the most central one. Whereas weak genetic structuring across the urbanization gradient did not allow us to formally test whether rates of immigration decreased relative to drift, towards more central urban populations, such a scenario may be especially apparent in large metropolitan cities where distances between core populations and potential demographic sources can be several orders of magnitude larger.
Notwithstanding the weak genetic differentiation, the RDA provided evidence of spatial genetic structure (and hence, dispersal limitation), even within the rural and within the (sub)urban cluster. Furthermore, the hierarchical spatial analysis clearly provided evidence that genetic drift is currently a more important driver of genetic structure than gene flow. This may reflect a reduction in gene flow compared with historical levels, a reduction in average local population sizes (and increase in genetic drift), or both. Whereas a small number of additional immigrants may be sufficient to replenish genetic variation and reduce inbreeding depression (‘genetic rescue’ sensu Ingvarsson, 2001), larger numbers of conspecifics are generally needed to positively affect population growth or vital rates and promote population stability (Taylor and Dizon, 1996) (‘demographic rescue’ sensu Brown and Kodric-Brown, 1977). Hence, demographic independence can be based on genetic independence, but not the other way around (Taylor and Dizon, 1999). Along these lines, a study of reproduction in British house sparrows has shown that, in 2 of 3 years, local recruitment in urban populations was insufficient to reach the predicted threshold required for population stability (Peach et al., 2008). As this demographic deficit was not counterbalanced by ample immigration from suburban or rural populations, this resulted in a population decline of 28% over the 3-year study period (Vincent, 2005). Unfortunately, estimates on genetic differentiation were not available to simultaneously measure genetic rescue effects.
Several other studies have quantified the genetic population structure of birds across gradients of urbanization. A study on common kestrels (Falco tinnuculus) did not reveal differences in levels of allelic richness or genetic differentiation between urban and rural areas (Riegert et al., 2010; Rutkowski et al., 2010). Likewise, pairwise levels of differentiation indicated weak structuring between urban and coastal populations of song sparrows (Melospiza melodia) (MacDougall-Shackleton et al., 2011). Yet, in both species and in house sparrows near the city of Ghent, levels of relatedness were consistently higher in urban populations (Riegert et al., 2010; MacDougall-Shackleton et al., 2011; Vangestel, 2011). Although such results could indicate subtle differences in connectivity between both areas, highly synchronized postnatal dispersal directions of siblings through parental control (Matthysen et al., 2010) or area-dependent levels of complete nest predation (MacDougall-Shackleton et al., 2011) might increase genetic similarity between individuals as well. In contrast, urban great tit (Parus major) populations showed higher genetic variation compared with forest populations, possibly indicating higher gene flow from urban parks to forests than vice versa. While levels of relatedness between urban great tits were also higher than expected based on random mating, comparative data of nearby forest populations are currently lacking (Björklund et al., 2010). Genetic variation across gradients of urbanization may reflect adaptive responses to heterogeneous environments evolved during the urbanization process, rather than differences in demographic features (Partecke and Gwinner, 2007; Badyaev et al., 2008; Evans et al., 2009). One of the most striking and best documented examples of such urban adaptation has been described in blackbirds (Turdus merula). Although originally a forest specialist, this species started to colonize urban environments during the early nineteenth century and showed a clear genetic basis for at least a number of behavioral and phenotypic trait shifts in urban populations (Partecke et al., 2006a, 2006b, Partecke and Gwinner, 2007). In free-ranging populations of house sparrows, Liker et al. (2008) showed that individuals from more urbanized areas were consistently smaller and in worse body condition than individuals from more rural ones. This difference in body mass remained significant when birds were housed in a common captive environment. Rather than reflecting reduced access to food for adults (for example, due to strong competition), or short-term responses to high food predictability (for example, by strategic mass regulation), Liker et al. (2008) considered this phenotypic variation to be under genetic control and possibly adaptive in urban environments. Genetic population connectivity, or lack thereof, has a central role in evolutionary ecological processes. Although evolutionary theory predicts high levels of gene flow to mitigate local adaptive responses (Hendry et al., 2001), they do not necessarily eliminate the adaptive potential of a population (Storz, 2005). For instance, non-random dispersal with respect to phenotypes has been held responsible for the evolution of differences in body mass in different avian populations (Garant et al., 2005; Storz, 2005; Postma and Van Noordwijk, 2005). Adaptive differences in body weight in house sparrows might persist under genetic admixture if smaller or lighter birds would either be forced or attracted to highly built-up areas and larger or heavier birds would tend to settle in more open areas. Although we cannot fully ascertain that the observed genetic structure in our study was caused by differences in demography across the urbanization gradient, we consider it unlikely to be driven by urban adaptations, as we have not yet identified phenotypic or behavioral patterns matching the urban-rural dichotomy. Indeed, as opposed to the study by Liker et al. (2008), urban and rural house sparrows did not differ in mean body size and condition in our study area. Urban individuals did show evidence of higher nutritional stress than individuals from less built-up areas, however, this variation was earlier shown to reflect constraints on home range behavior (Vangestel et al., 2010).
Earlier genetic studies on house sparrow populations were either conducted pre-decline or in non-urban environments. Hence, to our knowledge, this is the first study that provides a detailed description of the population genetic structure of contemporary house sparrow populations along an urbanization gradient after the onset of the decline. As pointed out in previous studies (Kekkonen et al., 2010), these results grant us the opportunity to monitor and evaluate future genetic changes in urban house sparrow populations and further improve conservation efforts by assembling and integrating our current knowledge on demography, behavior and population genetics in this species.
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad.kt145km5.
References
Anderson TR (2006). Biology of the Ubiquitous House Sparrow. Oxford University Press: New York, USA.
Badyaev AV, Young RL, Oh KP, Addison C (2008). Evolution on a local scale: developmental, functional, and genetic bases of divergence in bill form and associated changes in song structure between adjacent habitats. Evolution 62: 1951–1964.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004). GENETIX 4.05: Logiciel sous Windows TM pour la génétique des populations. Montpellier: Laboratoire Génome Populations Interactions.
Björklund M, Ruiz I, Senar JC (2010). Genetic differentiation in the urban habitat: the great tits (Parus major) of the parks of Barcelona city. Biol J Linn Soc 99: 9–19.
Borcard D, Legendre P, Drapeau P (1992). Partialling out the spatial component of ecological variation. Ecology 73: 1045–1055.
Brown JH, Kodric-Brown A (1977). Turnover rates in insular biogeography: effect of immigration on extinction. Ecology 58: 445–449.
Chamberlain DE, Toms MP, Cleary-Mcharg R, Banks AN (2007). House Sparrow (Passer domesticus) habitat use in urbanized landscapes. J Ornithol 148: 453–462.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999). New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153: 1989–2000.
Dawson DA, Horsburgh GJ, Krupa AP, Stewart IRK, Skjelseth S et al. (2012). Microsatellite resources for Passeridae species: a predicted microsatellite map of the house sparrow Passer domesticus. Mol Ecol Resour 12: 501–523.
Dawson DA, Horsburgh GJ, Küpper C, Stewart IRK, Ball AD, Durrant KL et al. (2010). New methods to identify conserved microsatellite loci and develop primer sets of high cross-species utility—as demonstrated for birds. Mol Ecol Resour 10: 475–494.
De Laet J, Summers-Smith JD (2007). The status of the urban house sparrow Passer domesticus in North-Western Europe: a review. J Ornithol 148: S275–S278.
Dias PC (1996). Sources and Sinks in Population Biology. Trends Ecol Evol 11: 326–330.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14: 2611–2620.
Evans KL, Gaston KJ, Sharp SP, McGowan A, Hatchwell BJ (2009). The effect of urbanization on avian morphology and latitudinal gradients in body size. Oikos 118: 251–259.
Ewens WJ (1972). The sampling theory of selectively neutral alleles. Theor Popul Biol 3: 87–112.
Excoffier L, Laval G, Schneider S (2005). An integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Falconer DS, MacKay TF (1996). Introduction to quantitative genetics. Longman: Essex, United Kingdom.
Fitzpatrick BM (2009). Power and sample size for nested analysis of molecular variance. Mol Ecol 18: 3961–3966.
Fleisher RC (1983). A comparison of theoretical and electrophoretic assessments of genetic structure in populations of the house sparrow (Passer domesticus). Evolution 37: 1001–1009.
Fortin MJ, Dale MRT (2005). Spatial Analysis: A Guide for Ecologists. Cambridge University Press: Cambridge, UK.
Gao H, Williamson S, Bustamante CD (2007). An MCMC approach for joint inference of population structure and inbreeding rates from multi-locus genotype data. Genetics 176: 1635–1651.
Garant D, Kruuk LEB, Wilkin TA, McCleery RH, Sheldon BC (2005). Evolutionary differentiation within a wild bird population caused by differential dispersal and genetic architecture. Nature 433: 60–65.
Gebremedhin B, Ficetola GF, Naderi S, Rezaei HR, Maudet C, Rioux D et al. (2009). Combining genetic and ecological data to assess the conservation status of the endangered Ethiopian walia ibex. Animal Conservation 12: 89–100.
Goudet J (1995). FSTAT (Version 1.2): A computer program to calculate F-statistics. J Hered 86: 485–486.
Griffith SC, Dawson DA, Jensen H, Ockendon N, Greig C, Neumann K et al. (2007). Fourteen polymorphic microsatellite loci characterized in the house sparrow Passer domesticus (Passeridae, Aves). Mol Ecol Notes 7: 333–336.
Griffith SC, IRK Stewart, Dawson DA, Owens IPF, Burke T (1999). Contrasting levels of extra-pair paternity in mainland and island populations of the house sparrow (Passer domesticus): Is there an 'island effect'? Biol J Linn Soc 68: 303–316.
Grubb TC (2006). Ptilochronology: Feather Time and the Biology of Birds. Oxford University Press, Oxford Ornithological Series: New York, USA.
Hanski I (1999). Habitat connectivity, habitat continuity, and metapopulations in dynamic landscapes. Oikos 87: 209–219.
Hawley DM, Hanley D, Dhondt AA, Lovette IJ (2006). Molecular evidence for a founder effect in invasive house finch (Carpodacus mexicanus) populations experiencing an emergent disease epidemic. Mol Ecol 15: 263–275.
Hedrick PW, Brussard PF, Allendorf FW, Beardmore JA, Orzack S (1986). Protein variation, fitness and captive propagation. Zoo Biol 5: 91–99.
Heij CJ (1985). Comparative ecology of the house sparrow Passer domesticus in rural, suburban and urban situations. PhD thesis Vrije Universiteit Amsterdam: Nederland.
Hendry AP, Day T, Taylor EB (2001). Population mixing and the adaptive divergence of quantitative traits in discrete populations: a theoretical framework for empirical tests. Evolution 55: 459–466.
Hole DG, Whittingham MJ, Bradbury RB, Anderson GQA, Lee PLM, Wilson JD et al. (2002). Widespread local house sparrow extinctions—agricultural intensification is blamed for the plummeting populations of these birds. Nature 418: 931–932.
Hole GD (2001). The population ecology and ecological genetics of the house sparrow Passer doemsticus on farmland in Oxfordshire. PhD thesis University of Oxford: UK.
Hurlbert SH (1971). The nonconcept of species diversity: a critique and alternative parameters. Ecology 52: 577–586.
Ingvarsson PK (2001). Restoration of genetic variation lost—the genetic rescue hypothesis. Trends Ecol Evol 16: 62–63.
Jakob EM, Marshall SD, Uetz GW (1996). Estimating fitness: a comparison of body condition indices. Oikos 77: 61–67.
Jakobsson M, Rosenberg NA (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Jombart T, Pontier D, Dufour AB (2009). Genetic markers in the playground of multivariate analysis. Heredity 102: 330–341.
Kalinowski ST (2004). Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5: 539–543.
Kekkonen J, Seppa P, Hanski I, Jensen H, Vaisanen RA, Brommer JE (2010). Low genetic differentiation in a sedentary bird: house sparrow population genetics in a contiguous landscape. Heredity 106: 183–190.
Keller LF, Jeffery KJ, Arcese P, Beaumont MA, Hochachka WM, Smith JNM et al. (2001). Immigration and the ephemerality of a natural population bottleneck: evidence from molecular markers. Proc R Soc Lond B 268: 1387–1394.
Liker A, Papp Z, Bokony V, Lendvai AZ (2008). Lean birds in the city: body size and condition of house sparrows along the urbanization gradient. J Anim Ecol 77: 789–795.
Luikart G, Cornuet J-M (1998). Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12: 228–237.
MacDougall-Shackleton EA, Clinchy M, Zanette L, Neff BD (2011). Songbird genetic diversity is lower in anthropogenically versus naturally fragmented landscapes. Conserv Genet 12: 1195–1203.
Matthysen E, Van Overveld T, Van de Casteele T, Adriaensen F (2010). Family movements before independence influence natal dispersal in a territorial songbird. Oecologia 162: 591–597.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Neigel JE (1997). A comparison of different alternative strategies for estimating gene flow from genetic markers. Ann Rev Ecol Syst 28: 105–128.
Neumann K, Wetton JH (1996). Highly polymorphic microsatellites in the house sparrow Passer domesticus. Mol Ecol 5: 307–309.
Paetkau D, Slade R, Burden M, Estoup A (2004). Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13: 55–65.
Parkin DT, Cole SR (1984). Genetic variation in the house sparrow, Passer domesticus, in the East-Midlands of England. Biol J Linn Soc 23: 287–301.
Partecke J, Gwinner E (2007). Increased sedentariness in European blackbirds following urbanization: a consequence of local adaptation? Ecology 88: 882–890.
Partecke J, Gwinner E, Bensch S (2006a). Is urbanisation of European blackbirds (Turdus merula) associated with genetic differentiation? J Ornithol 147: 549–552.
Partecke J, Schwabl I, Gwinner E (2006b). Stress and the city: urbanization and its effects on the stress physiology in European blackbirds. Ecology 87: 1945–1952.
Peach WJ, Vincent KE, Fowler JA, Grice PV (2008). Reproductive success of house sparrows along an urban gradient. Anim Conserv 11: 493–503.
Peakall R, Smouse PE (2006). Genalex 6: Genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Peery MZ, Beissinger SR, House RF, Berube M, Hall LA, Sellas A et al. (2008). Characterizing source-sink dynamics with genetic parentage assignments. Ecology 89: 2746–2759.
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004). GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95: 536–539.
Piry S, Luikart G, Cornuet J-M (1999). Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Postma E, Van Noordwijk AJ (2005). Gene flow maintains a large genetic difference in clutch size at a small spatial scale. Nature 433: 65–68.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pritchard JK, Wen W, Falush D (2007). Documentation for STRUCTURE Software: version 2.2. Available from http://pritch.bsd.uchicago.edu/software.
Prugnolle F, de Meeus T (2002). Inferring sex-biased dispersal from population genetic tools: a review. Heredity 88: 161–165.
Rannala B, Mountain JL (1997). Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94: 9197–9201.
Raymond M, Rousset F (1995). Genepop (version-1.2) - population-genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Reed DH, Frankham R (2003). Correlation between fitness and genetic diversity. Conserv Biol 17: 230–237.
Riegert J, Fainova D, Bystricka D (2010). Genetic variability, body characteristics and reproductive parameters of neighbouring rural and urban common kestrel (Falco tinnuculus) populations. Popul Ecol 52: 73–79.
Ronce O (2007). How does it feel to be like a rolling stone? Ten questions about dispersal evolution. Annu Rev Ecol Evol Syst 38: 231–253.
Rosenberg NA (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Rousset F (2008). Genepop ' 007: a complete re-implementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8: 103–106.
Rutkowski R, Rejt L, Tereba A, Gryczynska-Siemiatkowska A, Janic B (2010). Population genetic structure of the European kestrel Falco tinnunculus in Central Poland. Eur J Wildl Res 56: 297–305.
Sekercioglu CH, Ehrlich PR, Daily GC, Aygen D, Goehring D, Sandi RF (2002). Disappearance of insectivorous birds from tropical forest fragments. Proc Natl Acad Sci USA 99: 263–267.
Shaw LM, Chamberlain D, Evans M (2008). The house sparrow Passer domesticus in urban areas: reviewing a possible link between post-decline distribution and human socioeconomic status. J Ornithol 149: 293–299.
Stangel PW (1990). Genic differentiation among avian populations. Acta XX Congressus Internationalis Ornithologici, Christburg, New Zealand pp 2442–2453. New Zealand Ornithological Congress Trust Board, Wellington.
Storz JF (2005). Nonrandom dispersal and local adaptation. Heredity 95: 3–4.
Summers-Smith JD (1988). The Sparrows: A Study of the Genus Passer. T & AD Poyser: Calton, UK.
Taylor BL, Dizon AE (1996). The need to estimate power to link genetics and demography for conservation. Conserv Biol 10: 661–664.
Taylor BL, Dizon AE (1999). First policy then science: why a management unit based solely on genetic criteria cannot work. Mol Ecol 8: S11–S16.
ter Braak CJF, Šmilauer P (2002). Canoco 4.5. Microcomputer Power. Ithaca, New York.
Vallone PM, Butler JM (2004). Autodimer: a screening tool for primer-dimer and hairpin structures. BioTechniques 37: 226–231.
Van Loon EE, Cleary DFR, Fauvelot C (2007). ARES: software to compare allelic richness between uneven samples. Mol Ecol Notes 7: 579–582.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004). Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Vangestel C (2011). Relating phenotypic and genetic variation to urbanization in avian species: a case study on house sparrows (Passer domesicus). PhD thesis Ghent University: Belgium. ISBN 978-94-9069-571-2.
Vangestel C, Braeckman BP, Matheve H, Lens L (2010). Constraints on home range behaviour affect nutritional condition in urban house sparrows (Passer domesticus). Biol J Linn Soc 101: 41–50.
Vincent K (2005). Investigating the causes of the decline of the urban house sparrow Passer domesticus population in Britain. PhD thesis De Montfort University: UK.
Walsh PS, Metzger DA, Higuchi R (1991). Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques 10: 506–513.
Weir B (1990). Genetic Data Analysis: Methods for Discrete Population Genetic Data. Sinauer Associates: Sunderland, MA, USA.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wilson A (2004). The decline of the house sparrow. British Birds 97: 418–419.
Acknowledgements
We are grateful to T Burke for providing access to his lab facilities, H Matheve, T Cammaer and C Nuyens for field assistance and A Krupa and G Horsburgh for laboratory assistance. We are also indebted to R Butlin and two anonymous reviewers for insightful comments on an earlier draft of this manuscript. This study was financially supported by research project 01J01808 of Ghent University to LL. Genotyping was performed at the NERC Biomolecular Analysis Facility funded by the Natural Environment Research Council, UK. Fieldwork and genetic analyses were funded by research grants G.0149.09 of the Fund for Scientific Research—Flanders (to S Van Dongen and LL) and research grant 01J01808 of Ghent University (to LL). A Travel Grant of the Fund for Scientific Research—Flanders was provided to CV.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Vangestel, C., Mergeay, J., Dawson, D. et al. Genetic diversity and population structure in contemporary house sparrow populations along an urbanization gradient. Heredity 109, 163–172 (2012). https://doi.org/10.1038/hdy.2012.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.26
Keywords
This article is cited by
-
Genetic structure in neotropical birds with different tolerance to urbanization
Scientific Reports (2022)
-
Urbanization is associated with differences in age class structure in black-capped chickadees (Poecile atricapillus)
Urban Ecosystems (2021)
-
Contemporary and historical river connectivity influence population structure in western brook lamprey in the Columbia River Basin
Conservation Genetics (2019)
-
Increased differentiation between individuals, but no genetic isolation from adjacent rural individuals in an urban red squirrel population
Urban Ecosystems (2018)
-
Determining urban exploiter status of a termite using genetic analysis
Urban Ecosystems (2017)
