
Abstract
Geographic distributions of most temperate marine fishes are affected by postglacial recolonisation events, which have left complex genetic imprints on populations of marine species. This study investigated population structure and demographic history of European sprat (Sprattus sprattus L.) by combining inference from both mtDNA and microsatellite genetic markers throughout the species' distribution. We compared effects from genetic drift and mutation for both genetic markers in shaping genetic differentiation across four transition zones. Microsatellite markers revealed significant isolation by distance and a complex population structure across the species′ distribution (overall θST=0.038, P<0.01). Across transition zones markers indicated larger effects of genetic drift over mutations in the northern distribution of sprat contrasting a stronger relative impact of mutation in the species' southern distribution in the Mediterranean region. These results were interpreted to reflect more recent divergence times between northern populations in accordance with previous findings. This study demonstrates the usefulness of comparing inference from different markers and estimators of divergence for phylogeographic and population genetic studies in species with weak genetic structure, as is the case in many marine species.
Similar content being viewed by others


Introduction
Disentangling the evolutionary processes shaping population structure is of fundamental importance for understanding contemporary distributions of species and populations. Species distributions are in part determined by the environmental regimes within which a full life cycle can be sustained. However, environmental conditions change over time, potentially causing distributional shifts (for example, Perry et al., 2005) and may lead to isolation of demes from a previously panmictic population. Despite the usual lack of physical barriers in the sea it is now generally accepted that many marine organisms show population structures deviating from a pattern of panmixia and often distance may be the only factor restricting gene flow. Indeed, many species seem to display population structures reflecting barriers to gene flow over relatively small geographic scales (for example, Ruzzante et al., 1998; Bekkevold et al., 2005). Such genetic discontinuities are often referred to as phylogeographic breaks (Avise, 2000) and can arise and be maintained from a multitude of processes, including climatic and glacial cycles separating previously panmictic populations or connecting populations that diverged in allopatry (Barton and Hewitt, 1985). In the northern hemisphere, for example, population structures are highly influenced by the Quaternary glaciations (Maggs et al., 2008), presumably with the strongest imprint from the last glacial maximum (LGM) ∼20 000 bp. Furthermore, retention of juvenile stages by local oceanographic barriers has also been suggested to halt gene flow among contiguous populations (for example, Ruzzante et al., 1998). Lastly, genetic barriers may also be maintained by natural selection acting against migrants between locally adapted populations (that is, a ‘tension zone′ sensu Barton and Hewitt, 1985).
Throughout European waters, at least six major phylogeographic breaks or transition zones have been described for a variety of different marine taxa. These include first the area of the Aegean Archipelago and the Dardanelle Strait separating Black Sea from Mediterranean populations (Magoulas et al., 1996; Nikula and Vainola, 2003). Second, gene flow barriers separating the Adriatic from other eastern Mediterranean populations have also been reported (Stefanni and Thorley, 2003; Peijnenburg et al., 2006). Third, a genetic transition zone has been described in the Siculo-Tunisian strait and/or the Strait of Messina separating populations in western and eastern Mediterranean Basins (for example, Borsa et al., 1997; Rolland et al., 2007). Genetic transitions between the Atlantic–Mediterranean and between the Baltic–Atlantic regions are also pronounced across many taxa, with reports of clear breaks from the Strait of Gibraltar to the Almeria–Oran front (reviewed in Patarnello et al., 2007) and in the Skagerrak–western Baltic (reviewed in Johannesson and Andre, 2006). Lastly, the English Channel has also been identified to constitute a transition zone in the polychaete Pectinaria koreni (Jolly et al., 2005).
Few marine organisms are distributed throughout European continental waters, limiting the potential for conducting large-scale intraspecific comparisons of multiple transition zones. Phylogeographic studies have simultaneously spanned up to three of the above marine transition zones (e.g., Borsa et al., 1997; Rolland et al., 2007; Larmuseau et al., 2009), but few have comprised all of them for the same species (Nikula and Vainola, 2003; Wilson and Veraguth, 2010).
Here, we use the European sprat (Sprattus sprattus L.) as a model for studying contemporary population structure and distribution in relation to known transition zones. Sprat is a locally abundant, small pelagic clupeid fish with a nearly pan-European distribution: ranging from the Black Sea, along the northern Mediterranean and Iberian coasts to the Atlantic, North Sea, Norwegian coastal waters and into the Baltic Sea. Sprat thus occupies highly heterogeneous environments. A study using a mtDNA marker suggested a complex phylogeographic history with two major clades: one representing the clade that presumably colonised northern European waters following the LGM, and a second in the eastern Mediterranean and the Black Sea with a pre- or postglacial origin (Debes et al., 2008). A substructure was also evident within clades, with genetic differences within the ‘western’ clade between Atlantic–Baltic Sea and western Mediterranean populations, and within the ‘eastern’ clade between Adriatic Sea and Black Sea populations (Debes et al., 2008). A recent microsatellite study further demonstrated population structure across the Baltic–Atlantic transition zone (Limborg et al., 2009). In the current study, we analyse the combined data sets for mtDNA and microsatellite markers from the two above studies and also extend the previous sampling coverage. The combination of a new extensive sampling scheme with inference from both genetic markers allows us to gain insight into the underlying evolutionary mechanisms shaping population structure across multiple European transition zones. We then infer relative divergence times across transition zones, defined as ‘old’ (with a significant effect of mutation on genetic differentiation) vs ‘recent’ (with no significant effect of mutation on genetic differentiation), by contrasting effects of genetic drift and mutation for both marker types.
Mterials and methods
Samples
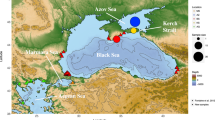
Samples covered the species’ distribution from its northern (Northeast Atlantic Ocean, North and Baltic Seas) to its southern (Mediterranean and Black Seas) range (Figure 1, Table 1). Sampling density, however, differed between north and south, reflecting a more continuous distribution of spawning locations in the north, compared with the south where major populations presumably are presently restricted to the Gulf of Lion, the northern Adriatic Sea and the Black Sea Basins (Debes et al., 2008). Additional occurrences in estuarine areas around the Iberian Peninsula have been reported, but populations are thought to be in strong decline or even disappeared (Cabral et al., 2001). Findings in the northern Aegean Sea have been reported (Deval et al., 2002), but our own sampling efforts in that region have not been successful. Assumed occurrences in the Strait of Sicily have never been confirmed (O Jarboui, personal communication). Data from a total of 21 sampling stations representing 19 locations were included in the analysis (Figure 1). Of these, mtDNA variation was reported for seven locations in Debes et al. (2008). Microsatellite data were compiled for 17 sampling stations: 11 stations as reported by Limborg et al. (2009) and six additional stations extending the previous northerly dominated coverage southwards via the English Channel and the Atlantic into the Mediterranean and eastwards into the Black Sea (Figure 1). This data set effectively increased sampling coverage throughout the species distribution including the Mediterranean region. Samples were, with few exceptions, collected on spawning sites during the spawning season, which differs among populations. Temporal replicates were collected from two locations (see Table 1, Debes et al. (2008) and Limborg et al. (2009) for more details on sampling).

Samples analysed for microsatellites (black squares with three letter sample ID) and mtDNA (white circles with two-letter sample ID) markers. Sample ID corresponds to Table 1. Underlined samples represent locations with temporally repeated sampling. All samples analysed with the mtDNA marker (white circles) are the same as in Debes et al. (2008). Grey-shaded areas with labels in italics show transition zones separating the Baltic and Atlantic (including the North Sea, abbreviated Balt–Atl), Atlantic and Mediterranean (Atl-WMed), western and eastern Mediterranean (WMed-EMed) and the Mediterranean and Black Sea (EMed-Black), respectively. See text and Table 1 for more details.
Populations and geographic transition zones studied
In the following, we refer to all geographic zones with observed genetic discontinuities as ‘genetic transition zones’, regardless of the underlying mechanisms. Of the six major cross-species transition zones defined a priori, the English Channel does not appear to constitute one for sprat, as samples on either side of the English Channel show spatial as well as temporal genetic homogeneity (Limborg et al., 2009; Glover et al., 2011). We could not address a potential transition zone between the Adriatic Sea and the eastern Mediterranean as no sprat could be obtained from the eastern Mediterranean. In the current study, we are thus obliged to use the Adriatic population for investigating genetic differentiation between the eastern Mediterranean and the Black Sea. We investigated genetic differentiation across the remaining four major transition zones separating the following regions: (i) the Baltic Sea from the Atlantic region (here, the latter includes the North Sea and English Channel, Balt–Atl), (ii) the Atlantic region from the Mediterranean Sea (Atl–WMed), (iii) the western from the eastern Mediterranean Sea (WMed–EMed), and (iv) the eastern Mediterranean Sea from the Black Sea (EMed–Black; Figure 1).
Molecular analyses
Samples from seven locations were genotyped for both mitochondrial and nuclear DNA markers, of which both marker types were analysed for the same individuals in three of the samples (Table 1). Thus, four samples were typed only for mtDNA and 14 only for microsatellite markers.
In total, 210 individuals from seven locations (Table 1) were sequenced for a partial fragment of the 5′-end of the mitochondrial control region, as described in Debes et al. (2008).
A total of 1531 individuals, including 556 new to this study, were typed for nine species-specific microsatellite loci: Spsp47D, Spsp77C, Spsp133, Spsp155, Spsp170, Spsp202, Spsp219, Spsp256 and Spsp275 (Dailianis et al., 2008). DNA extraction and PCR amplification were performed as described in Dailianis et al. (2008). PCR-amplified microsatellite fragments were analysed either on a BaseStation 51 DNA fragment analyser (MJ Research, Skovlunde, Denmark) followed by semi-automatically typing of genotypes with the software CARTOGRAPHER 1.2.6 (MJ Geneworks Inc., Skovlunde, Denmark) (samples GOT, GDA, BOR05, BOR06, ARK, BEL, KAT, SKA, GER04, GER05, ENC, CEL, BoB and ADR), or on a Beckman Coulter CEQ 8000 (Beckman-Coulter, Fullerton, CA, USA) automated sequencer (samples SKA, LIO, BLW and BLE). For the latter, allele sizes were scored with the software CEQ 8000 Genetic Analysis System (version 8.0.52; Beckman-Coulter). All individual runs included a 400-bp ladder (Applied Biosystems, Foster City, CA, USA; Beckman-Coulter). To obtain consistency in genotype scoring among runs and between platforms, we (i) analysed from two to four heterozygote control individuals spanning the anticipated allelic ranges, (ii) double-typed two samples (n=40) on both platforms, and (iii) split the SKA sample into two groups of n=50 and genotyped on different platforms to test for consistency in allele frequency estimates between platforms (see Supplementary File S1 for further details on validation of scoring consistency).
Genetic variation
For microsatellites, potential effects of technical or sampling artefacts were assessed by checking for effects of null alleles and departure from Hardy–Weinberg Equilibrium (HWE) and gametic phase equilibrium (LD) using MICRO-CHECKER 2.2.3 (Van Oosterhout et al., 2004) and GENEPOP 4.0 (Raymond and Rousset, 1995), respectively. In all following analyses including multiple tests, results were corrected with the sequential Bonferroni method (Rice, 1989). Overall genetic variation and diversity were estimated by allelic richness (Ar) for each sample and locus using FSTAT 2.9.3 (Goudet, 1995). Weir and Cockerham′s inbreeding coefficient θ IS (Weir and Cockerham, 1984) was estimated for each locus and sample using FSTAT 2.9.3. Numbers of alleles (A), expected and observed heterozygosity (HE and HO, respectively) were calculated for all loci and samples using Arlequin 3.5 (Excoffier and Lischer, 2010).
Outlier analysis
Potential effects of natural or hitchhiking selection on microsatellite loci may obscure inferred patterns of neutral demographic processes (Nielsen et al., 2006). We tested for any such patterns using BayeScan 1.0, following the Bayesian method described in Foll and Gaggiotti (2008). To obtain sufficient convergence of MCMC chains, we ran 10 pilot runs of 5000 iterations and an additional burn-in of 5 × 106 iterations with a thinning interval of 50 and a final sample size of 50 000. For comparison, we also used the model by Excoffier et al. (2009b) as implemented in Arlequin 3.5 (Excoffier and Lischer, 2010) by running 10 000 simulations.
Inference of total number of populations
To infer the number of populations in our samples we analysed the microsatellite data using the Bayesian clustering model implemented in STRUCTURE 2.3.1 (Pritchard et al., 2000). This model infers population structure by clustering individual multilocus genotypes into a given number of populations (K) by minimising LD and overall departure from HWE. We used the admixture model with correlated allele frequencies among populations. We initially considered five trials for each value of K from one to ten. To ascertain adequate convergence of the MCMC model we used a burn-in of 5 × 105 iterations, followed by 2 × 106 sampled iterations. We considered the mean probability values of lnP(X|K) given by the programme, as well as the ΔK method (Evanno et al., 2005) to infer the most likely number of populations. For subsequent biological interpretations of K we focused on the smallest value capturing most of the structure in the data, as suggested in the manual. Subsequently, we repeated the analysis on subsets of major clusters detected by the first run, to detect potential finer scale substructure. All analyses were performed with either no population information, or including population sample as prior information, according to Hubisz et al. (2009). The latter model has been shown to outperform the original model for clustering populations at weak structure (that is, FST values <0.10) and with limited numbers of microsatellite markers (Hubisz et al., 2009).
Statistical analyses of overall population structure
We used Arlequin 3.5 to estimate pairwise FST from mtDNA haplotype frequencies (using conventional F-statistics based on haplotype frequencies only) between all samples, and compared these to the pairwise ΦST estimates (that is also based on genetic distances among haplotypes) reported in Debes et al. (2008).
Owing to a denser coverage for samples analysed with microsatellites, the description of population structure was mainly based on these markers. We thus estimated an overall and pairwise genetic differentiation using Weir and Cockerham′s (1984) estimator (here, referred to as θST) and 95% confidence intervals (CI) using the approach described in Neff and Fraser (2010). Statistical significance of pairwise θST estimates was tested using permutation tests implemented in FSTAT 2.9.3. RSTCALC 2.2 (Goodman, 1997) was used to estimate pairwise RST between all samples and significance was tested by 1000 permutations, whereas 95% CI were obtained by bootstrapping 1000 times over loci. A principal component analysis (PCA), based on allele frequencies, for all 17 population samples was performed using PCAGEN 1.3.1 (available at: www2.unil.ch/popgen/softwares/pcagen.htm). Significance of each principal component (PC) was tested by 10 000 randomisations.
To test if the geographic pattern of genetic differentiation is caused by isolation by distance we ran Mantel tests for pairwise matrices between geographic distance and genetic distance in Arlequin 3.5 with 100 000 permutations. This was performed for both marker types and for both measures of genetic differentiation separately (linearised equivalents of FST and ΦST for mtDNA, FST and RST for microsatellites, respectively). Geographic distance was estimated by direct shipping distance between coordinates of sampling locations calculated with the programme Netpas Distance (Netpas).
Demographic effects on population structure
Spatial population expansions are expected to result in higher population-specific FST values in marginal populations that have potentially undergone more founder events and received fewer immigrants than populations closer to an ancestral source population (Foll and Gaggiotti, 2006; Gaggiotti and Foll, 2010). To statistically test a potential effect of range expansion on population-specific differentiation, we used GESTE v2.0 (Foll and Gaggiotti, 2006) to estimate population-specific FST values following the approach by Balding and Nichols (1995). Depending on the underlying demographic history of the species, this FST estimator describes the differentiation of each population from the overall meta-population (under a migration-drift model), or from a common ancestral source population (under a fission model) (Foll and Gaggiotti, 2006).
Genetic differentiation across transition zones
Subsequent analyses focused on genetic patterns across four transition zones (Figure 1), synthesising results from the two marker types. For mtDNA data, we pooled samples fulfilling the criteria of not crossing a transition zone as well as not showing statistically significant differentiation for either the FST or ΦST pairwise estimates within the regional groups (Table 1, also see Supplementary File S2 for pairwise ΦST and FST). Applying this approach, groups of samples thus represented the following five regions: the Baltic Sea (abbreviated BALT in Table 1), the Northeast Atlantic (incl. North Sea; ATLA), the western Mediterranean (WMED), the Adriatic Sea (ADRI) and the Black Sea (incl. Strait of Bosporus; BLAS). Similarly, for microsatellite data we pooled subsets of samples showing no statistically significant pairwise θST, to represent the same five regions (Table 1). Samples from within the Baltic–Atlantic transition zone (BEL, KAT and SKA; Figure 1) and a single sample from the Celtic Sea (CEL) that showed weak, but significant, differentiation from neighbouring samples (Supplementary File S3) were omitted from this analysis to avoid potential confounding effects from pooling non-panmictic populations.
The programme POWSIM 4.0 (Ryman and Palm, 2006) was used to evaluate statistical power of both types of markers for detecting pairwise genetic differentiation at FST levels ranging from 0.00 to 0.10. The programme simulates the divergence of two to several subpopulations from a single ancestral population through genetic drift to a given overall FST value defined by controlling effective population size (Ne) and number of generations (t). To best reflect the assumingly large Ne of sprat, we let Ne=10 000 and varied t from 0 to 2078 for simulating different levels of differentiation. After the simulation, each subpopulation was sampled at n=80 and divergence from genetic homogeneity was tested with Fisher′s exact test. This procedure was repeated 1000 times and the proportion of significant outcomes was used to estimate statistical power for detecting pairwise genetic differentiation. Founder events in populations of more recently colonised areas may have left a stronger imprint from genetic drift, resulting in higher levels of pairwise FST between neighbouring populations. To infer our power for detecting such events we tested four scenarios corresponding to observed genetic differentiation between populations on both sides of the four studied transition zones. Specifically, for the Baltic–Atlantic transition zone, we pooled samples from the BALT and ATLA groups (Table 1) to represent allele frequencies for the ancestral population at the onset of the simulation process. Similarly, we pooled samples for the groups flanking each of the remaining transition zones (Table 1, Figure 1). For the mtDNA analysis, we only pooled the two geographically closest samples on each side of a transition zone, as including more samples led to violation of the maximal number of alleles (or haplotypes) for a given marker (50) allowed by POWSIM, owing to a large number of private haplotypes in all samples.
Pairwise FST estimates based on mtDNA haplotype frequencies are expected to be mainly shaped through genetic drift, at least on time scales where mutations can be largely ignored. In contrast, the ΦST estimator takes the number of mutational differences among haplotypes into account and is able to reveal higher resolution on divergence time between populations having accumulated specific mutations over time (see Excoffier et al., 1992 for more details). Comparisons of FST and ΦST estimates for mtDNA sequences across multiple transition zones is therefore expected to reveal relative imprints from genetic drift and mutation in explaining the level of genetic differentiation. Thus, we repeated analyses in Arlequin 3.5 using 20 000 permutations to obtain pairwise estimates of FST (using conventional F-statistics) and ΦST (using a distance matrix based on haplotype nucleotide differences corrected with the base substitution model of Tamura and Nei (1993)) between the regions represented by the five major groups described above.
The relative effects of genetic drift and mutations in explaining genetic differentiation across transition zones were also examined for microsatellite data by applying the RST permutation test in SPAGeDi 1.2 (Hardy and Vekemans, 2002). The test compares observed RST values based on allele size differences assuming a stepwise mutation model (SMM) with a corresponding frequency distribution (ρRST) obtained by randomly permuting over allelic states following an infinite allele model of mutation. A significantly larger observed RST implies a significant role of mutation for explaining population structure and suggests that divergence occurred over very long time scales (Pons and Petit, 1996; Hardy et al., 2003). Significance was tested with 20 000 permutations using a one-sided test (RST>ρRST) (Slatkin, 1995). Similar tests were applied for global RST estimates for each locus and all loci together. For locus Spsp275, a total of four individuals from the ENC and BoB samples had considerably larger alleles (50–200 bp longer) than the maximum sizes observed in all other samples. These (rare) alleles may be the results of one or more insertion events, and including them would violate the assumption of a SMM. Information for these four individuals was therefore ignored in RST permutation tests.
Results
One microsatellite locus (Spsp154) failed to amplify consistent fragment lengths between the two genotyping platforms in the 40 calibration individuals and was discarded from further analyses. For the remaining eight loci, scoring of genotypes was consistent between the genotyping platforms (see Supplementary File S1 for more information on calibration results).
Overall genetic variation at mtDNA
For mtDNA, a total of 128 different haplotypes with 82 segregating sites were observed in the seven samples (Debes et al., 2008). Haplotype diversity (h) for each sample is reported in Table 1.
Overall genetic variation at microsatellite loci
A total of 64 individuals with more than two missing genotypes were excluded, leaving 1467 individuals for which 99.97% of all loci were scored successfully (all summary statistics for each locus and sample are reported in Supplementary File S4). MICRO-CHECKER suggested the potential presence of null alleles for 35 (out of 136) sample locus pairs, and stutter-prone scoring at eight sample locus pairs. However, no general trends of a specific locus or sample were evident and subsequent analyses including or excluding information from affected loci did not change results. After correcting for multiple tests, significant deviations from HWE remained for 10 of 136 (7%) tests distributed among four loci (Spsp275: 3 significant tests, Spsp219: 1, Spsp133: 4 and Spsp170: 2) (Supplementary File S4). One out of 28 locus pairs showed significant LD (Spsp219, Spsp133). However, this was only observed in four of the 17 population samples. A similar test for LD by Limborg et al. (2009) on a subset of these samples did not show overall LD for any of these loci, and LD is thus not expected to incur a general bias in our analyses. Nuclear genetic diversity assessed by Ar is reported for each sample in Table 1.
Outlier analysis
The BayeScan test indicated three outlier loci (Spsp170, Spsp202 & Spsp275) potentially subject to divergent selection, whereas the test implemented in Arlequin supported this only for the latter two loci, which also showed the highest level of support for divergent selection (Supplementary File S5). Thus, to test for potential effects of the two outlier loci found by both methods we conducted all subsequent analyses using (i) all loci, (ii) excluding each of the two outlier loci and (iii) excluding both outlier loci.
Estimation of the total number of populations
The global Bayesian clustering analysis revealed the highest likelihood for models with K=3 and 4, whereas the ΔK method suggested K=3 (Figure 2a) irrespective of whether prior sample information was used or not. Visual inspection revealed that setting K>3, did not add further meaningful inference (not shown), and we only show results for K=3, as this presumably captures the major biological structure across samples (that is, population clusters representing, respectively, the Baltic Sea, the Atlantic region (including the North Sea), and the Mediterranean region (including the Black Sea)) (Figure 2b). Subsequent analyses comprising either samples from within the Baltic Sea, the Atlantic region or both, with and without prior sample information did not reveal further substructuring (data not shown). When including prior sample information, an analysis comprising Mediterranean and Black Sea samples revealed substructuring (K=3) with the Gulf of Lion (LIO), the Adriatic (ADR) and the Black Sea (BLW and BLE) samples presumably representing genetically distinct populations (Figure 2c). The tuning parameter, r, for the latter model ranged from 0.06 to 0.12 among the five replicate runs. Values of r below 1.00 indicate that ancestry proportions differ among sampling locations and that the inclusion of prior sample information significantly increased the power for detecting weak population structure (Hubisz et al., 2009). Altogether, five clusters could hence be detected using Bayesian clustering (Figure 2).

(a) Probability of each tested potential number of populations (K) inferred from the mean probability value ln [P(X|K)] (white squares) and the ΔK method (black squares) (see text for more details). (b) Individual population membership plotted for K=3. (c) Individual population membership when repeating the cluster analysis for the Mediterranean samples (LIO, ADR, BLW and BLE) for K=3 and including prior information of sample location.
Population structure and demography
Estimators of pairwise mtDNA differentiation (FST and ΦST) revealed significant population differentiation in most comparisons (see below and Supplementary File S2). Microsatellites also revealed highly significant population structure, with an overall θST of 0.038 (95% CI=0.015 to 0.064, P<0.001) and pairwise θST estimates ranging between 0.001 and 0.100. Genetic differentiation between temporal samples from both the Bornholm Basin (BOR) and the German Bight (GER) was low and non-significant (θST<0.005), suggesting temporal stability of the observed spatial structure in these regions. The level of genetic structure varied among different geographical regions with mostly non-significant estimates of pairwise θST within major oceanic basins in contrast to comparisons among basins (Supplementary File S3). In the PCA, the first two PCs explained a significant proportion of the total genetic variance (PC1 and PC2, P<0.001; PC3 to PC10, P=1.000). PC1 explained 49.1% of the total genetic variance and grouped samples corresponding to the two previously described major phylogenetic clades separated at the western and eastern Mediterranean Sea transition zone, with further separation of LIO from all other samples (Figure 3). Samples across the Baltic–Atlantic transition zone showed a clear East–West trend along PC2 (21.8%). This overall pattern remained significant although the level of differentiation was reduced when excluding the two outlier loci (not shown).

Genetic relationships of samples as revealed from the two first PCs from the microsatellite-based PCA. Sample IDs correspond to Figure 1.
Isolation by distance was highly significant for both FST (R2=0.82) and RST (R2=0.89) for all microsatellite loci (Table 2). When only excluding one of the two outlier loci, results remained significant but with levels of explained variance reduced by 12–19% (Table 2). When simultaneously excluding both outlier loci the explained variation was reduced more drastically by 63–66% for both FST and RST, and only the FST-based test remained significant (R2=0.30; Table 2). For mtDNA, a significant but weaker pattern of isolation by distance was revealed only for the ΦST values (R2=0.31).
Population-specific FST estimates showed an increasing trend from the west (Atlantic Ocean) eastward into both the Baltic Sea in the north and into the Adriatic and Black Seas in the south (Figure 4). This overall pattern remained when excluding one or both outlier loci, although FST values reduced to 0.003–0.027 when excluding all outlier loci.

Population-specific FST values for microsatellite markers with black vertical bars representing mode values and grey boxes illustrating the 95% highest probability density interval (the smallest interval that contains 95% of the values). Oceanic region is given for each sample next to the vertical axis and correspond to names in Figure 1 and Table 1.
Genetic differentiation across transition zones
Overall, the power to detect genetic differentiation owing to allelic drift did not vary significantly across the four transition zones (Supplementary File S6). The mtDNA marker data lacked sufficient statistical power for detecting values of FST<0.02 but could reliably detect levels of differentiation above this level (FST=0.02; power=0.872–0.952). The eight microsatellites exhibited adequate power for detecting true FST>0.005 (0.998–1.00). Type-one errors (FST=0) did not seem to seriously violate an assumed 5% α-level for either type of marker used (Supplementary File S6).
For the mtDNA data, pairwise FST and ФST estimates crossing one or more of the three southern transition zones were significant for both estimators, but with considerably higher values of the ΦST estimator (Figure 5). One striking disparity, however, was observed across the northern Baltic–Atlantic transition zone where the Baltic group (BALT) showed statistically significant differentiation from the Atlantic group (ATLA) for the drift-based FST estimator but not for the ΦST estimator.

Pairwise FST (white bars) and ΦST (black bars) estimates for mtDNA sequences across transition zones with samples pooled into the following regions; Baltic Sea (BALT), Atlantic Ocean (ATLA), western Mediterranean (WMED), Adriatic Sea (ADRI) and the Black Sea (BLAS) (see text for more details). Pairwise comparisons between regions directly connected by each of the four transition zones are denoted with abbreviations in italic corresponding to Figure 1. All estimates are significantly >0 (α=0.05) unless denoted with ns.
The mtDNA results were supported by the microsatellite-based analyses including all loci where the mutation-based RST estimator was not significantly higher than the purely drift-based ρRST distribution between the Baltic (BALT) and Atlantic (ATLA) groups, suggesting a negligible mutational imprint across this transition zone (Figure 6a). Also for microsatellite markers, mutations appeared to have had a relatively larger role in genetic differentiation across southern transition zones, evidenced by a significant pattern of RST>ρRST in seven comparisons (Figure 6a). A non-significant effect was observed between the Adriatic (ADRI) and Black Sea (BLAS) groups (Figure 6a). When excluding either of the two outlier loci an overall pattern of a strong mutational effect across southern transition zones remained as three and five tests remained significant when excluding Spsp202 and Spsp275, respectively (Figure 6b). Conversely, no tests were significant when excluding both outlier loci simultaneously (Figure 6d).

Observed microsatellite-based pairwise RST point estimates (black squares) between regional groups and the ρRST distribution (open squares and vertical bars represent mean and 95% CI) obtained by randomly permuting over allelic states (see text for more details). Genetic differentiations are shown across all transition zones as described for figure 5. Asterisks denote comparisons where allele size differences (that is, mutation) inferred from RST explain a significant part of the genetic differentiation. Results are shown for tests including all loci (a), excluding: Spsp202 (b), Spsp275 (c) and both Spsp202 and Spsp275 (d).
Discussion
By combining inference from mitochondrial and nuclear DNA markers we gained new insights into the potential effects of historical demography in explaining distribution-wide population structure of sprat covering four major transition zones. Both marker types showed clear regional patterns of population structure and especially microsatellites indicated a pattern of isolation by distance. The mtDNA marker successfully inferred old from more recent divergence times across the different transition zones. The advantage of combining multiple marker types has previously been demonstrated in marine fishes (for example, Gonzalez and Zardoya, 2007; Wilson and Veraguth, 2010; Andre et al., 2011). However, to our knowledge the present study is the first to directly compare relative imprints from genetic drift and mutation between markers and throughout the geographic distribution of a small pelagic marine fish.
Overall population structure and historical demography
The initial STRUCTURE analysis identified three population clusters and corroborated previous assertions about genetically isolated populations in the Atlantic region, the Mediterranean and Baltic Sea, respectively, using fewer samples (Limborg et al., 2009). In this study, a subsequent analysis of population subsets revealed a finer structure within the Mediterranean/Black Sea region (Figure 2c). The eastern Black Sea sample (BLE) appears admixed with the Adriatic Sea population (ADR; Figure 2c), which could be explained by contemporary gene flow, shared ancestry or homoplasy. However, these explanations appear unlikely considering the intermediate location of the non-admixed sample (BLW) and this result more likely reflects analytical limitations of the method when few markers are applied (Hubisz et al., 2009). Overall, the clustering result is in accordance with pairwise mtDNA and microsatellite differentiation estimates, which also revealed the highest genetic discontinuities among major oceanic basins (Supplementary Files S2 and S3) corresponding to the five clusters detected by STRUCTURE (that is, the Baltic, Atlantic, western Mediterranean, Adriatic and Black Seas).
When using STRUCTURE with the six neutrally behaving microsatellite loci only, no structure was detected (most likely K=1), suggesting that the observed population structure is largely driven by the presumed outlier loci Spsp202 and Spsp275 (data not shown). This raises the question of whether the results indeed reflect the demographic history of the species, however, at least two facts speak in favour of this. First, the overall population structure is supported by independent analyses of both mitochondrial and nuclear DNA, and second, a PCA and estimates of pairwise θST omitting the two outlier loci detected a similar and statistically significant (albeit weaker) pattern of population structure. Moreover, the resolving power of STRUCTURE tends to be low with few markers at low divergence (Hubisz et al., 2009). Lastly, increased power is expected for detecting low genetic differentiation between predefined populations based on pairwise tests comparing allele frequencies (like θST), compared with STRUCTURE, which does not consider such a priori-defined subgroups (Pritchard et al., 2007).
Our results thus support a pattern with at least five more or less reproductively isolated genetic clusters in sprat throughout its distribution. Similar levels of clustering are reported for other small pelagic fishes (for example, Bekkevold et al., 2005; Grant, 2005; Gonzalez and Zardoya, 2007), albeit those studies spanned narrower geographic regions. Although our focus here is the large-scale distribution, we cannot rule out the potential existence of non-sampled locally isolated populations at smaller geographic scales. Indeed, a recent study has shown existence of population structure between Norwegian fjord populations and the North Sea sprat population (Glover et al., 2011), suggesting the existence of isolated local populations.
The grouping of genetic clusters along PC1 in the PCA (Figure 3) corresponds with three distinct phylogenetic clades occurring in; (i) the Atlantic region (including the Baltic Sea), (ii) the western Mediterranean and (iii) the eastern Mediterranean (including the Black Sea). This pattern suggests that historical and phylogeographic patterns also explain a significant part of neutral genetic variation at microsatellites in combination with contemporary migration-drift processes. An effect of range expansions on genetic variation was supported by population-specific FST values which were in agreement with a ‘fission model’ where populations expanded from west (the Atlantic Ocean) into the Mediterranean and the Black Sea, as well as into the North and Baltic Seas. Such a demographic model was further supported by the significant patterns of isolation by distance, where especially the differentiation revealed by microsatellites was explained by geographic distance. When excluding both outlier loci, the FST-based pattern of isolation by distance was still apparent, whereas the RST-based pattern, however, became non-significant. This latter observation may indicate that for genetic differentiation at microsatellites indeed contemporary migration-drift processes may be more important than mutations, since differential mutations among populations with low or no gene flow would most likely result in larger RST estimates and significant isolation by distance. However, this result may also simply reflect technical issues if for example, neutrally behaving microsatellites are more constrained in size, which would deflate true RST values at these loci. Fragment size (alleles) distributions, however, did not suggest such a pattern in our data (not shown), thus we cannot further assess this potential explanation. Alternatively, the RST estimate may exhibit larger variance than FST (Balloux and Lugon-Moulin, 2002) explaining the observed non-significant isolation by distance pattern for RST when two outlier loci were removed.
For many marine organisms in the Northeast Atlantic, major refugia during the LGM included regions south of the Bay of Biscay with potential smaller inter-glacial refugia further north (Maggs et al., 2008). For example, the thornback ray (Raja clavata L.) presumably persisted in at least two Atlantic refugia along the Iberian Peninsula and the Azores (Chevolot et al., 2006). A similar scenario of north- and eastward expansions from one or more south westerly Atlantic refugia for sprat cannot be ruled out and would be in accordance with our results. The northwards range expansion most likely happened after the LGM in accordance with the biogeographical history of the Baltic Sea, which did not support the present-day marine fauna before ∼9–7000 bp (Sohlenius et al., 2001). Together with the study by Debes et al. (2008), our findings of large mutational differences at the mtDNA marker suggest old population divergence across the Mediterranean transition zones potentially pre-dating the LGM. A similar scenario of pre-LGM divergence within the Mediterranean has also been suggested for another fish species (Wilson and Veraguth, 2010).
Differentiation of marginal populations
High microsatellite-based population-specific FST values and slightly reduced mtDNA haplotype diversity of Adriatic Sea and Black Sea populations compared with Atlantic samples (Figure 4, Table 1) point to a relatively old split between an eastern Mediterranean and a western Mediterranean/Atlantic clade. Debes et al. (2008) explained the present-day pattern at the southern edge of the distribution of sprat in the Mediterranean as a result of northwards shifting isotherms since the LGM. Populations in the northernmost Mediterranean basins occur at their physiological limit and likely represent trapped remnants of a formerly more widespread core population in the Mediterranean.
However, under this scenario, the separation in the Mediterranean of an eastern and western clade might also reflect local founder events from cryptic inter-glacial refugia pre-dating the LGM. Postglacial colonisation of the Black Sea could also, in theory, have taken place from a refugial population now only represented in this area. Moreover, the observed FST pattern (Figure 4) could also be consistent with a stepping-stone model with lower migration rates (and higher drift) for marginal populations, without inference about the directionality of founder events (Gaggiotti and Foll, 2010).
A post hoc permutation test in FSTAT revealed reduced allelic richness, Ar, in the Baltic group (BALT; Ar=15.06±0.10 (mean±s.e.)) compared with the Atlantic group (ATLA; Ar=20.23±0.14 (mean±s.e), one-tailed test, P<0.001), consistent with observations for the mtDNA (h) (Table 1). Similar tests did not reveal significantly reduced Ar in the Adriatic Sea or Black Sea populations compared with the Atlantic group (P>0.11). Assuming that the distribution of sprat populations follows a stepping-stone pattern; an alternative, but not mutually exclusive, explanation for reduced diversity and increased differentiation in the marginal Baltic Sea population can be due to reduced immigration of new alleles compared with more ‘central’ populations. At first sight a similar explanation appears incongruent with the relatively higher Ar in the marginal Adriatic population (Table 1). One explanation for this could be that higher microsatellite mutation rates and longer time since presumably older founder events have erased signals of reduced genetic diversity. However, strong signatures from old founder events would not be expected if contemporary immigration is the dominating factor for shaping genetic diversity in marginal populations. For example, increased environmental stress in marginal populations may reduce immigration into locally adapted populations leading to reduced diversity and greater differentiation of these populations (Excoffier et al., 2009a). Lastly, congruent patterns of reduced genetic diversity in Baltic populations of other ‘classical’ marine fishes (reviewed in Johannesson and Andre, 2006) are suggestive of a general trend reflecting shared founder histories, reduced immigration, environmental adaptation and/or other unknown factors simultaneously reducing Ne in this marginal sea.
Disentangling effects of genetic drift and mutation across transition zones
We found indications that both genetic drift and mutation explain genetic differentiation across transition zones, but the relative effect of each varied among the different transition zones studied. This result is likely to reflect population splitting events at different time scales. More recently diverged populations will resemble each other in terms of haplotypes and alleles present, as fewer new mutations are expected to have accumulated. Pairwise differentiation between the Baltic and Atlantic groups revealed a significant FST and a lower non-significant ΦST for the mtDNA, together with a non-significant RST test for microsatellites. Genetically admixed populations within this transition zone (Figure 2) could suggest on-going gene flow eroding signals from population-specific mutations. Alternatively, recent divergence between Baltic and Atlantic populations may explain the lack of detectable differentiation in this transition zone. Although these two explanations may not be mutually exclusive, this, together with the geologic history of the Baltic region, reinforces the notion of the Baltic Sea maintaining the most recently established sprat population among those studied (see above).
Interestingly, we see a pattern of generally larger effects from mutation in most pairwise comparisons spanning one or more of the three southern transition zones. This is in accordance with the results from Debes et al. (2008) pointing towards relatively deep splits between samples within the Mediterranean region reflecting no or very little gene flow in combination with large divergence times. Most comparisons crossing the transition zone separating the western and eastern Mediterranean Sea, suggested by clade analysis to represent the deepest phylogeographic split (Debes et al., 2008), show an accordingly larger effect from mutation and isolation. However, for microsatellites this result was mainly explained by two loci also exhibiting outlier behaviour, and thus, potentially violating the assumptions of neutrality. Furthermore, increased frequency of alleles affected by positive selection may lead to deviations from the neutral allele distribution expected under a SMM (see below). As a consequence, the mtDNA-based results may better reflect true differences between genetic drift and mutation here. The somewhat reduced mutation effect at the mtDNA between the Adriatic Sea population and the Black Sea samples (Figure 5) is in accordance with expected shorter divergence times within the two major clades and/or higher levels of gene flow (Debes et al., 2008). The mtDNA-based estimators of genetic drift (FST) and mutational distance (ΦST) were both significant between the Adriatic and Black Sea groups, as opposed to across the transition zone separating the Baltic and Atlantic populations. This suggests an intermediate divergence time between the Adriatic Sea and Black Sea groups. The relatively high mutational effect between the Atlantic and western Mediterranean groups within the ‘western’ clade also suggest a considerably older divergence between these groups than between the Atlantic and Baltic Sea groups. However, owing to lack of Atlantic samples south of the Bay of Biscay (presumably reflecting low densities), we cannot rule out possible confounding effects from a potential undetected structure around the Iberian Peninsula. In such a case, a sample from the more southern population would be more appropriate when testing differentiation across the Atlantic–western Mediterranean transition zone owing to a potentially more recent shared ancestry with the western Mediterranean population.
A large effect of mutation relative to drift, in combination with restricted gene flow, should lead to a genome-wide pattern of RST>ρRST. The significant contribution of mutations in explaining differentiations among the major clades with microsatellites was mainly driven by the two loci Spsp202 and Spsp275 (Supplementary File S7), which were also suggested to be affected by directional selection (Supplementary File S5). Thus, great caution should be taken when quantitatively inferring effects from mutation and neutral genetic drift when using these loci. Nevertheless, the congruent results observed for two independent loci may suggest a biological meaningful pattern of increased mutational effects on microsatellite variation across southern transition zones. We, however, cannot rule out that these outlier loci behave in a non-neutral fashion and thus violate the SMM model leaving the microsatellite-based results inconclusive. Alternatively, our results may have indicated a general trend of microsatellite loci mainly reflecting more recent migration-drift processes as suggested from significant patterns of isolation by distance, whereas genetic variation at mtDNA markers appeared better suited for inferring older demographic histories. Indeed, for the mtDNA results we did find varying relative effects of genetic drift and mutation across different transition zones indicative of varying divergence times between different sprat populations.
Other statistical methods offer more direct estimates of bottlenecks, time since divergence and gene flow. However, the signal in our data is likely too weak to obtain reliable estimates from these analyses, as testified by the fact that analyses using the approaches of Piry et al. (1999) and Garza & Williamson (2001) were inconclusive of past bottlenecks in any of our populations (not shown). Also, attempts to apply IMa (Hey and Nielsen, 2004) on mtDNA and microsatellite data to infer divergence times and gene flow resulted in non-converging MCMC chains, reflecting a lack of information in the data. Instead, by taking advantage of a more indirect approach of comparing relative imprints from mutation and genetic drift, we were able to distinguish putatively ‘old’ from more ‘recent’ population divergence across transition zones putatively characterised by varying levels of gene flow. We thus expect that this approach may be useful in other applications for organisms characterised by weak structure due to recent divergence, large Ne and/or high gene flow.
Data Archiving
Data have been deposited at Dryad: doi:10.5061/dryad.m247bg66.
References
Andre C, Larsson LC, Laikre L, Bekkevold D, Brigham J, Carvalho GR et al. (2011). Detecting population structure in a high gene-flow species, Atlantic herring (Clupea harengus): direct, simultaneous evaluation of neutral vs putatively selected loci. Heredity 106: 270–280.
Avise JC (2000). Phylogeography: The History and Formation of Species. Harvard University Press Cambridge, MA.
Balding DJ, Nichols RA (1995). A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96: 3–12.
Balloux F, Lugon-Moulin N (2002). The estimation of population differentiation with microsatellite markers. Mol Ecol 11: 155–165.
Barton NH, Hewitt GM (1985). Analysis of Hybrid Zones. Annu Rev Ecol Syst 16: 113–148.
Bekkevold D, Andre C, Dahlgren TG, Clausen LAW, Torstensen E, Mosegaard H et al. (2005). Environmental correlates of population differentiation in Atlantic herring. Evolution 59: 2656–2668.
Borsa P, Blanquer A, Berrebi P (1997). Genetic structure of the flounders Platichthys flesus and P. stellatus at different geographic scales. Mar Biol 129: 233–246.
Cabral HN, Costa MJ, Salgado JP (2001). Does the Tagus estuary fish community reflect environmental changes? Clim Res 18: 119–126.
Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL (2006). Phylogeography and population structure of thornback rays (Raja clavata L., Rajidae). Mol Ecol 15: 3693–3705.
Dailianis T, Limborg M, Hanel R, Bekkevold D, Lagnel J, Magoulas A et al. (2008). Characterization of nine polymorphic microsatellite markers in sprat (Sprattus sprattus L.). Mol Ecol Resour 8: 861–863.
Debes PV, Zachos FE, Hanel R (2008). Mitochondrial phylogeography of the European sprat (Sprattus sprattus L., Clupeidae) reveals isolated climatically vulnerable populations in the Mediterranean Sea and range expansion in the northeast Atlantic. Mol Ecol 17: 3873–3888.
Deval MC, Ates C, Bök T, Oray IK (2002). Investigations of the distribution of the eggs and larvae of the sprata Sprattus sprattus L. 1758, in the Sea of Marmara. ICES CM 2002/L:27 (www.Ices.dk).
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Foll M, Petit RJ (2009a). Genetic consequences of range expansions. Annu Rev Ecol Evol S 40: 481–501.
Excoffier L, Hofer T, Foll M (2009b). Detecting loci under selection in a hierarchically structured population. Heredity 103: 285–298.
Excoffier L, Lischer HEL (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among dna haplotypes - application to human mitochondrial-DNA restriction data. Genetics 131: 479–491.
Foll M, Gaggiotti O (2006). Identifying the environmental factors that determine the genetic structure of Populations. Genetics 174: 875–891.
Foll M, Gaggiotti O (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180: 977–993.
Gaggiotti OE, Foll M (2010). Quantifying population structure using the F-model. Mol Ecol Resour 10: 821–830.
Garza JC, Williamson EG (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Glover K, Skaala Ø, Limborg M, Kvamme C, Torstensen E (2011). Microsatellite DNA reveals population genetic differentiation among sprat (Sprattus sprattus) sampled throughout the Northeast Atlantic, including Norwegian fjords. ICES J Mar Sci 68: 2145–2151.
Gonzalez EG, Zardoya R (2007). Relative role of life-history traits and historical factors in shaping genetic population structure of sardines (Sardina pilchardus). BMC Evol Biol 7: 197.
Goodman SJ (1997). Rst Calc: a collection of computer programs for calculating estimates of genetic differentition from microsatellite data and determining their significance. Mol Ecol 6: 881–885.
Goudet J (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Grant WS (2005). A second look at mitochondrial DNA variability in European anchovy (Engraulis encrasicolus): assessing models of population structure and the Black Sea isolation hypothesis. Genetica 125: 293–309.
Hardy OJ, Charbonnel N, Freville H, Heuertz M (2003). Microsatellite allele sizes: A simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482.
Hardy OJ, Vekemans X (2002). SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hey J, Nielsen R (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760.
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332.
Johannesson K, Andre C (2006). Life on the margin: genetic isolation and diversity loss in a peripheral marine ecosystem, the Baltic Sea. Mol Ecol 15: 2013–2029.
Jolly MT, Jollivet D, Gentil F, Thiebaut E, Viard F (2005). Sharp genetic break between Atlantic and English Channel populations of the polychaete Pectinaria koreni, along the North coast of France. Heredity 94: 23–32.
Larmuseau MHD, Van Houdt JKJ, Guelinckx J, Hellemans B, Volckaert FAM (2009). Distributional and demographic consequences of Pleistocene climate fluctuations for a marine demersal fish in the north-eastern Atlantic. J Biogeogr 36: 1138–1151.
Limborg MT, Pedersen JS, Hemmer-Hansen J, Tomkiewicz J, Bekkevold D (2009). Genetic population structure of European sprat Sprattus sprattus: differentiation across a steep environmental gradient in a small pelagic fish. Mar Ecol Prog Ser 379: 213–224.
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J et al. (2008). Evaluating signatures of Glacial Refugia for North Atlantic Benthic Marine Taxa. Ecology 89: S108–S122.
Magoulas A, Tsimenides N, Zouros E (1996). Mitochondrial DNA phylogeny and the reconstruction of the population history of a species: the case of the European anchovy (Engraulis encrasicolus). Mol Biol Evol 13: 178–190.
Neff BD, Fraser BA (2010). A program to compare genetic differentiation statistics across loci using resampling of individuals and loci. Mol Ecol Resour 10: 546–550.
Nielsen EE, Hansen MM, Meldrup D (2006). Evidence of microsatellite hitch-hiking selection in Atlantic cod (Gadus morhua L.): implications for inferring population structure in nonmodel organisms. Mol Ecol 15: 3219–3229.
Nikula R, Vainola R (2003). Phylogeography of Cerastoderma glaucum (Bivalvia: Cardiidae) across Europe: a major break in the Eastern Mediterranean. Mar Biol 143: 339–350.
Patarnello T, FAMJ Volckaert, Castilho R (2007). Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographical break? Mol Ecol 16: 4426–4444.
Peijnenburg KTCA Fauvelot C, Breeuwer AJ, Menken SBJ (2006). Spatial and temporal genetic structure of the planktonic Sagitta setosa (Chaetognatha) in European seas as revealed by mitochondrial and nuclear DNA markers. Mol Ecol 15: 3319–3338.
Perry AL, Low PJ, Ellis JR, Reynolds JD (2005). Climate change and distribution shifts in marine fishes. Science 308: 1912–1915.
Piry S, Luikart G, Cornuet JM (1992). BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Pons O, Petit RJ (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144: 1237–1245.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pritchard J K, Wen X, Falush D (2007) Documentation for structure software: Version 2.2 Available from http://pritch.bsd.uchicago.edu.
Raymond M, Rousset F (1995). Genepop (Version-1.2) - population-genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rolland JL, Bonhomme F, Lagardere F, Hassan M, Guinand B (2007). Population structure of the common sole (Solea solea) in the Northeastern Atlantic and the Mediterranean Sea: revisiting the divide with EPIC markers. Mar Biol 151: 327–341.
Ruzzante DE, Taggart CT, Cook D (1998). A nuclear DNA basis for shelf- and bank-scale population structure in northwest Atlantic cod (Gadus morhua): Labrador to Georges Bank. Mol Ecol 7: 1663–1680.
Ryman N, Palm S (2006). POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6: 600–602.
Slatkin M (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Sohlenius G, Emeis KC, Andren E, Adren T, Kohly A (2001). Development of anoxia during the Holocene fresh-brackish water transition in the Baltic Sea. Mar Geol 177: 221–242.
Stefanni S, Thorley JL (2003). Mitochondrial DNA phylogeography reveals the existence of an Evolutionarily Significant Unit of the sand goby Pomatoschistus minutus in the Adriatic (Eastern Mediterranean). Mol Phylogenet Evol 28: 601–609.
Tamura K, Nei M (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Weir BS, Cockerham CC (1984). Estimating F-Statistics for the analysis of population-structure. Evolution 38: 1358–1370.
Wilson AB, Veraguth IE (2010). The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol Ecol 19: 4535–4553.
Acknowledgements
This study is a contribution of the EU Network of Excellence MARBEF, Marine Biodiversity and Ecosystem Functioning. The study was carried out with financial support from the European Commission through the FP6 projects UNCOVER (contract no. 022717) and RECLAIM (contract no. 044133). It does not necessarily reflect the views of the European Commission and in no way anticipates the Commission′s future policy in this area. Financial support was also partly provided by the BaltGene project funded by BONUS Baltic Organisations’ Network for Funding Science EEIG, and the Linnaeus Centre for Marine Evolutionary Biology at University of Gothenburg (www.cemeb.science.gu.se). Thomas Damm Als is thanked for help with illustrations. Two anonymous referees provided constructive comments and suggestions on an earlier draft. Lastly, we are deeply grateful to Michele Casini, Georgiy Shulman, Atilla Özdemir, Melek Işinibilir, Pascal Lorance, Arnaud Souplet, Andrea Ramšak, Rudi Voss and Yves Verin for invaluable help with providing samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary material accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Limborg, M., Hanel, R., Debes, P. et al. Imprints from genetic drift and mutation imply relative divergence times across marine transition zones in a pan-European small pelagic fish (Sprattus sprattus). Heredity 109, 96–107 (2012). https://doi.org/10.1038/hdy.2012.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.18
Keywords
This article is cited by
-
Chaotic genetic structure and past demographic expansion of the invasive gastropod Tritia neritea in its native range, the Mediterranean Sea
Scientific Reports (2020)
-
Influence of hydrodynamic connectivity on the genetic structure and gene flow of the common pandora Pagellus erythrinus
Hydrobiologia (2019)
