
Abstract
The history of population size and migration patterns leaves its mark in the genetics of populations. We investigate the genetic structure of the edible frog, Pelophylax esculentus in the Danish archipelago and adjacent countries. This frog is of particular interest because it is a hybrid that, in this area, forms all-hybrid populations of diploid (LR) and triploid (LLR and LRR) genomotypes with no (or very few) adults of the parental species (LL and RR). This study is the first to cover the entire geographic range of Danish, Swedish and German all-hybrid populations, documenting their extent and providing a broad picture of their diversity of neutral genetic markers and genomotype proportions. With 18 microsatellite markers, we found that genetic diversity declines northwards in agreement with the glacial refuge and central-marginal hypotheses; however, populations on small and medium-sized islands are no less diverse than those on large islands and continental peninsulas. Isolation by distance exists across the archipelago with limited influence of fragmentation by brackish seawater. The extremely low genetic diversity in all-hybrid populations, compared with adjacent populations, may be responsible for the maintenance of their special breeding system. We also show large variation among ponds in proportions of LLR, LR and LRR genomotypes, but little geographic pattern in their distribution. Instead, we found relationships between the genomotype proportions and some of 15 habitat parameters monitored. Body size differences among LLR, LR and LRR further suggest ecological differences.
Similar content being viewed by others

Introduction
Genetic structure arises when restrictions on population size and gene flow lead to loss of genetic diversity, because finite populations lose genetic diversity due to genetic drift (Wright, 1931). Thus, genetic diversity is expected to decrease with distance from a glacial refuge as a consequence of sequential founder events (post-glacial colonization hypothesis) and/or from a species’ distribution centre to its periphery due to decreased density and increased isolation (central-marginal hypothesis; Eckert et al., 2008). Furthermore, mainland populations have been shown to be more diverse than island populations, with genetic diversity decreasing with island size (Frankham, 1996). In accordance with the island size effect, human-mediated habitat fragmentation reduces neutral genetic diversity of populations worldwide (Aguilar et al., 2008).
Genetic structure is of concern from a conservation perspective, because genetic variation is the prerequisite for adaptation to new and changing environments (Frankham, 1996). Even in stable environments, inbreeding depression can lower the fitness of small and isolated populations (Keller and Waller, 2002). On the other hand, genetic structuring can lead to speciation, as evidenced by the much higher proportions of endemic species per area on islands than on the mainland (Kier et al., 2009).
The archipelago, adjacent peninsulas and continent of Denmark, southern Sweden and northern Germany offers an opportunity for studying the effect of natural fragmentation and island size on the genetic structure of low-mobility terrestrial and freshwater species. The whole area was a coherent land mass 9200–7000 years ago, when the current herpetofauna colonized Scandinavia from the south or southeast (Fog et al., 1997). Sequential post-glacial founder events should thus be detectable as decreasing genetic diversity in a northern direction, whereas founder population sizes should be proportional to present-day island sizes and not to their degree of isolation. Depending on the relative strengths of genetic signals from, first, the dilution effect from post-glacial migration and, second, the drift effect related to island size and isolation, both signals might be detectable in low-mobility species at microsatellite marker loci.
Amphibians are attractive subjects for population genetic studies partly because of their limited mobility, which leads to population structure over relatively short distances, and partly because of their dramatic population declines worldwide (Beebee, 2005). This study was made on the edible frog, Pelophylax esculentus (called Rana esculenta until Frost et al., 2006). This frog is of particular interest to evolutionary biologists because of its hybrid origin and bizarre reproductive mode. Moreover, Denmark, southern Sweden and northern Germany comprise the only known larger area where these frogs form biologically unique all-hybrid populations of diploid and triploid hybrids in the absence of the two parental species (Graf and Polls Pelaz, 1989; Christiansen et al., 2005; Arioli, 2007, Chapters 3 and 4).
P. esculentus is widespread in Europe and is a hybrid between the pool frog, P. lessonae, and the lake frog, P. ridibundus. The genomic constitutions, here called genomotypes as in Bogart et al. (2007), of these parental species are denoted LL and RR, respectively. Hybrids use different hybridogenetic modes of reproduction in different areas reviewed by Graf and Polls Pelaz (1989). In most of Europe, P. esculentus frogs have the diploid genomotype LR and reproduce hemiclonally, transmitting a clonal copy of only the R genome to gametes. They thus depend on backcrossing with R. lessonae to form a new generation of hybrids (L-E system). Inter-hybrid matings usually result in RR offspring that are inviable because they are homozygous for recessive deleterious mutations in the clonally transmitted R genome (Vorburger, 2001a). Modifications of this system include reproduction with P. ridibundus instead of P. lessonae in eastern Europe (R-E system), triploid LLR and LRR individuals (can be present in all breeding systems) and the all-hybrid populations (E-E system).
Studies on gamete patterns in all-hybrid populations agree that triploid frogs of both sexes produce haploid gametes with the genome they have in double dose, whereas diploid frogs produce LR or R gametes (Graf and Polls Pelaz, 1989; Jakob, 2007, Chapter 5; Christiansen, 2009). The majority of LR gametes are eggs, as LR sperm is rare in most populations. As the male-determining Y factor is not present in R genomes, LRR males are probably only formed when rare LR sperm fertilizes R eggs. This explains why LRR males are very infrequent in most ponds (Christiansen, 2009).
Although the genetics of reproduction in all-hybrid P. esculentus is now largely understood, two central questions about all-hybrid populations remain. The first question is how these populations remain all-hybrid. Various studies have shown that non-hybrids (LL and RR) arise from hybrid × hybrid matings, but die off during larval stages (Christiansen et al., 2005; Arioli, 2007, Chapter 3). The inviability of RR offspring from hybrid × hybrid matings in the L-E system arises from the diploid hybrids’ clonal mode of reproduction (Vorburger, 2001a), but this explanation cannot hold for all-hybrid populations where recombination between the two Ls occurs in LLR frogs and between the two Rs in LRR frogs (Christiansen and Reyer, 2009). An alternative hypothesis is that genetic diversity in the L and R genomes within all-hybrid populations is so low that, despite regular recombination within them, LL and RR offspring nevertheless become homozygous for a sufficient number of recessive deleterious mutations to kill them. To test this hypothesis, the genetic diversity of all-hybrid populations should be compared with that of adjacent, mixed populations.
The second question concerns why all-hybrid populations differ in genomotype proportions (LLR, LR and LRR). Several studies have documented differences in the proportions of LLR, LR and LRR males and females among ponds (Christiansen et al., 2005; Jakob, 2007, Chapter 2). In contrast, models have shown that differently composed founder populations should all converge on the same equilibrium genomotype proportions, unless among-pond variation in the gametogenetic pattern or in selection on the various genomotypes is present (Som and Reyer, 2006; Christiansen, 2009). The relative importance of gamete patterns and ecology for shaping genomotype proportions are, however, not known. An analysis of the abundance of different genomotype proportions and their distribution over geographic areas and habitats would help to elucidate factors responsible for differentiation in genomotype proportion.
This study is the first to cover the entire geographic range of Danish, Swedish and German all-hybrid populations, documenting their extent and providing a broad picture of diversity at neutral genetic markers and genomotype proportions. We analysed 13L- and 13R-specific microsatellite loci and expected genetic diversity to increase: (1) toward the south and perhaps also east for the R genome, because P. ridibundus has its distribution centre southeast of the study area; (2) from small over large islands to the mainland and (3) in mixed populations compared with all-hybrid populations, to explain the inviability of LL and RR in the latter. Moreover, we investigate population differentiation, isolation by distance and the effect of sea on isolation by distance. Finally, we relate proportions of the LLR, LR and LRR genomotypes to geography and habitat.
Materials and methods
Frog sampling
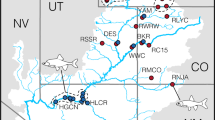
Frogs were collected in 118 ponds in six countries (Figure 1; Table 1, pond coordinates in Supplementary Information 1). The frogs were caught at night by hand or dip net while dazzling them with a strong torch. The goal was to obtain at least 30 adult frogs per pond, including at least 10 adults of each sex, but this was not always accomplished (sample sizes in Supplementary Information 1). All frogs had a finger tip clipped for DNA analysis and were returned to their home pond. Danish, Swedish and German frogs were also measured from snout to vent (with straight back) and sexed by the presence or absence of vocal sac openings. Frogs with snout-to-vent lengths of ⩾55 mm (⩾50 mm for P. lessonae), when measured with a slide caliper with an accuracy of 0.1 mm, were defined as adults. All ponds were sampled only once, except for the Swedish ponds, which were sampled twice in 2005; recaptures were excluded from the second sample.

Map based on Fog et al. (1997); Rybacki and Berger (2001); Zeisset and Beebee (2001); Arioli (2007, Chapter 5) and our information. L-L, L-E and L-E-R are breeding systems reported in the literature.
Pond data
Geographic coordinates were obtained from Google Earth (Google 2009, http://earth.google.com) and Det Levende Danmarkskort (Kort & Matrikelstyrelsen, 2002).
In Danish and Swedish ponds, 15 habitat parameters were measured between May and July 2005. Oxygen content, oxygen saturation, pH, connectivity and water temperature were measured at night, between 00:20 and 06:40 hours, 10 cm below the surface 0.5–2 m from the shore with a HQ20 LDO sensor and a Hach sensIon pH electrode (Hach-Lange GmbH, Hegnau, Switzerland). The remaining 10 parameters were recorded by daylight. Surface area was calculated from the estimated length and width of the pond at the visit, taking the shape into account. Depth was measured as the maximum depth a fist-sized stone sunk when lowered from a small rubber boat. Submerged/floating vegetation and emerging vegetation were scored as the percentage of the surface area covered. Bank shape was scored as 1 (flat), 2 (medium) or 3 (steep). The presence of short grass (cut or grazed), long grass, bushes/single trees, wood and agriculture within 100 m of the ponds was each quantified on an ordinal scale from −100 to +100. Positive values describe the percentage of the pond periphery lined by the given land vegetation type, whereas negative values described the distance in meters to the vegetation type, if it did not touch the pond. Land vegetation types not present within 100 m of the pond were given the score −100. Time and date of the registrations were noted.
German ponds were categorized only as open land, intermediate or forest ponds.
Genomotype determination
Frogs from all Danish, Swedish, ‘Germany Inland’ and seven of the Baltic ponds were analysed with our full set of 18 microsatellite primer pairs (listed in Supplementary Information 2 and 3). Extraction, PCR and sequencer protocols are described in Christiansen and Reyer (2009), except that forward primer Ga1a19redisigned (Christiansen, 2009) was used instead of Ga1a19.
With very few exceptions (see Results) all alleles encountered were specific to either the L or R genome (Supplementary Information 2 and 3). Specificity was known for many alleles from earlier studies (Christiansen et al., 2005; Jakob, 2007, Chapter 1; Christiansen, 2009), confirmed here in P. lessonae and P. ridibundus, and inferred for newly sampled alleles in P. esculentus on the basis of alleles with known specificity. Of the eight primer pairs that amplify both L and R-specific alleles, four show dosage effect. At each of these four loci, the relative intensity of L and R alleles amplified was used to distinguish LLR, LR and LRR frogs (Christiansen et al., 2005). Genomotype was determined as that indicated by the majority of these dosage effect loci, and the remaining loci were checked for congruence with this genomotype.
Whereas LLLR and LRRR tetraploids can be identified by extreme dosage effects (Christiansen et al., 2005), LLRR tetraploids will appear as LR diploids if homozygous for all L and for all R loci (Jakob, 2007, Chapter 1). To estimate the magnitude of this potential error source, the probability of homozygosity at all L and at all R loci was, for each pond, calculated as the product of 1−HE of all loci. Partly heterozygous LLRR frogs should show LR dosage effect in combination with incongruent heterozygosity for some L and/or R loci.
All frogs from ‘Germany Coast’ and Bornholm, and eight of the Baltic state ponds had their genomotypes determined by Martina Arioli and Christian Jakob on the basis of three to four of the dosage effect loci mentioned above. Allele data from these frogs were not used, because they did not comprise the full set of 18 loci. To keep sample sizes relatively constant in the genetic analyses, allele data from the second sample in Swedish ponds were also not used.
Allele data
The full allele genotype of frogs, for which allele data were obtained (Table 1), was inferred on the basis of their overall genomotype. However, sometimes the number of alleles at one or few loci was, upon repeated analyses, incongruent with the overall genomotype (Supplementary Information 3 presents multilocus genotypes for all frogs, incongruent loci are highlighted and our handling of them illustrated). When a locus was incongruent in only one or two individuals in a pond, the phenomenon was deemed quantitatively unimportant and the locus was coded as missing data in these frogs. However, when a locus showed incongruence in more than two individuals in a pond, the phenomenon was attributed to either local genome inspecificity or to a null allele. In cases of local genome inspecificity, the allele in question was ascribed to the L genome in some frogs and to the R genome in other frogs in accordance with the overall genomotype. In case of a null allele, its frequency was assessed in hemizygous individuals where non-amplification was not masked by other alleles. Assuming the same frequency of null alleles in hemizygous and non-hemizygous frogs, null alleles were randomly added to apparently homozygous frogs. This correction procedure should be unproblematic, as further analyses were made only on allele frequencies; not on observed homo- and heterozygosities.
Genetic parameters were calculated in SPAGeDi, version 1.2 g (Hardy and Vekemans, 2002), which accepts a mixture of individuals with different ploidy levels. L and R genome data were split into separate input files, because SPAGeDi does not accept different ploidy levels in the same animal (in an LLR frog, the L is diploid and the R is haploid). HE (expected heterozygosity, that is genetic variability corrected for sample size; Nei, 1978), FST (a measure of population differentiation; Weir and Cockerham, 1984) and DS (Nei's standard genetic distance; Nei, 1978) were calculated for use in the analyses described below.
Analyses with allele data
Genetic diversity was measured as expected heterozygosity, HEL and HER, and analysed for effects of breeding system (all-hybrid or mixed populations), latitude, longitude and land area rank. Tests were done in R, version 2.9.0 (R Development Core Team, 2009). HE was compared between breeding systems using Mann–Whitney U test (performed as Wilcoxon tests with unmatched pairs in R), because HEL for all-hybrid populations was not normally distributed. HE was correlated with latitude, longitude and land area rank using Spearman rank correlations because most of the parameters were not normally distributed, and because of the difficulty of determining relevant land areas for peninsulas and continents. Land area ranks were based on land size and connectivity, so that Danish islands were ranked according to size (ranks 1–14) followed by the peninsulas Sweden (rank 15) and Jutland (rank 16) and the continental German+Baltic areas (rank 17). Locality 23a+b was regarded as an island and 9a and 9b as separate islands.
Isolation by distance was investigated as the correlation between pairwise FST/(1–FST) and ln(km) and between DS and ln(km). Pairwise geographic distances were calculated with Geographic Distance Matrix Generator, version 1.2.3 (Ersts, American Museum of Natural History, Center for Biodiversity and Conservation, http://biodiversityinformatics.amnh.org/open_source/gdmg) from geographic coordinates after conversion to decimal degrees on the homepage of the Federal Communications Commission (http://www.fcc.gov/mb/audio/bickel/DDDMMSS-decimal.html). Correlations were tested with Mantel tests with 1 00 000 permutations in the program zt, version 1.1 (Bonnet and Van de Peer, 2002). The influence of sea on isolation by distance was tested with partial Mantel tests (also with 1 00 000 permutations in zt) using a sea matrix as a second explanatory variable. In the sea matrix, 0 coded for land connection between ponds, 0.5 for a bridge and 1 for sea. The relative proportions of total variance explained by ln(km) and the sea matrix were calculated using ANOVAs in R.
Analyses with genomotype proportions
The samples were frequently biased towards one sex, possibly due to differential behaviour of males and females. Therefore, male and female genomotype proportions were treated separately in all analyses involving genomotype proportions.
For visualizing genomotype proportions, ternary plots (triangle diagrams) were drawn in the programme Past, version 1.91 (Hammer et al., 2001). Genomotype proportions were based on at least 10 individuals of the same sex; smaller samples were not included in the diagrams.
To test whether genomotype proportion differences increased with geographic distance, pairwise differences in genomotype proportion between ponds were calculated as their Euclidian distances in the ternary plots and then correlated with ln(km) by Mantel tests, as described above. Ternary coordinates were converted into XY coordinates following Chen et al. (2007).
The effects of latitude, longitude and their interaction on genomotype abundances were analysed with logistic regressions in R. Logistic regressions handle proportional data taking sample sizes into account and thus make exclusion of small samples unnecessary. A model including latitude, longitude and their interaction was fitted separately to each genomotype within sex. As the data exhibited overdispersion with the binomial error distribution (residual deviation >the degrees of freedom), quasibinomial error distributions and F tests were used.
The analysis of the effect of habitat parameters on genomotype proportions was done on 62 Danish and Swedish ponds. Seven ponds (2a, 4z, 14a, 21b, 23a, 23b, 138) were excluded because of missing data, whereas ‘ponds’ 17a and 25c were assemblages of 3 and 2 ponds, respectively, that entered the analysis with separate habitat and genomotype proportion data. Oxygen content, oxygen saturation, pH, connectivity and water temperature were corrected for both the time and date of measurement, as these were thought to have influenced the parameters. Correction was done by first converting time to decimal numbers after midnight and dates to the number of days after 1st May. Then residuals were extracted from a multiple regression of each parameter on time and date and their interaction. Surface area, depth, both types of pond vegetation and short and long grass were corrected for date in the same way.
To reduce the 15 potentially correlated variables to a smaller number of independent factors, we subjected them to a correlation matrix-based principal component analysis (PCA; SYSTAT version 11) with an orthogonal varimax rotation. Principal components (PCs) with an eigenvalue ⩾1.00 were retained and tested for relations with genomotype proportions using logistic regressions with quasibinomial error distributions as described above.
The impact of forest on German ponds was analysed with logistic regressions with quasibinomial error distributions and with the categories open land, intermediate and forest as a continuous variable with values 1, 2 and 3, respectively. Swedish ponds were included in a second analysis, whereas too few Danish forest ponds were sampled for inclusion of Danish ponds.
Means are reported with±1 standard deviation.
Results
Microsatellite data
All alleles could be assigned to either the L or R genome with low levels of incongruence (see Supplementary Information 2 for microsatellite length ranges, number of alleles found, HE and incongruence, and Supplementary Information 3 for multilocus genotypes). With 13L and 13R microsatellite loci analysed in 2507 frogs, 476 cases (1.05%) of locus incongruence with the consensus genomotype were encountered. The majority of incongruent cases were German and Baltic, although the majority of samples were Danish and Swedish. In 11 ponds, incongruence was attributed to allele inspecificity; in 21 ponds to null alleles. Only one case of incongruence could possibly be attributed to tetraploidy. This adult frog from the Swedish pond 001 showed LR dosage at all dosage effect loci but showed heterozygosity for L alleles and homozygosity for an R allele at another locus. The probability of mistaking LLRR for LR without signs of incongruence was only 0.042±0.117, averaged over Danish, Swedish and German ponds (no polyploidy in Baltic ponds).
Breeding system distribution
Fifty-four Danish (excluding Bornholm), 12 Swedish and 33 German ponds were investigated in the supposedly all-hybrid area (Figure 1; genomotype frequencies in Supplementary Information 1). The 1457 adult Danish frogs were hybrids without exception. Of the Swedish frogs, 725 were hybrids whereas 4 (0.4%) were LL. The German sampling area included some of the transition zone from all-hybrid to other population types in the south, as the ponds Po, Ha2, Ha6 and Her had high proportions of parental species (71.4, 12.5, 33.3 and 63.0%, respectively). The remaining 30 German ponds were all-hybrid or almost so. In these ponds, 624 adult frogs were hybrid, whereas 3 (0.5%) were LL. Thus, we confirmed that the large area of Denmark, southern Sweden and northern Germany is inhabited by all-hybrid populations.
Samples from eastern Bornholm were dominated by LRR in both sexes. Other genomotypes were LLR, LR and RR (R-E system); however, RR constituted only 0.45% of the males compared with 26.1% of females.
All frogs from the Baltic States were diploid, except for one LLL individual from EE-H. The northernmost water frog populations in the Baltic States were pure P. lessonae, as is also the case in Sweden, Norway and a now-extinct native population in the United Kingdom (Zeisset and Beebee, 2001; Arioli, 2007, Chapter 5). A little further south, P. lessonae and P. esculentus occurred together (diploid L-E system), whereas P. ridibundus only occurred in samples from Riga (LV-J) and southwards.
Genetic diversity
Genetic diversity was investigated in Denmark, Sweden, Germany Inland and half of the Baltic ponds, where the full microsatellite data set was available (Figure 2; HE and n per pond in Supplementary Information 1). Within the all-hybrid ponds, HE in the L genome ranged from 0.000 to 0.351; mean 0.157±0.074. In the R genome, HE was generally higher, ranging from 0.000 to 0.521; mean 0.186±0.117. Outside the all-hybrid area, HEL averaged 0.315±0.129 in four German ponds and 0.286±0.122 in seven Baltic ponds (Figure 2). HER averaged 0.453±0.060 in the four German ponds and was 0.615 in the single Baltic pond where calculation was possible. HE was thus significantly lower in all-hybrid populations than in mixed populations in Germany (Mann–Whitney U tests: L genome: W=31, P=0.008; R genome: W=10, P=0.002) and the Baltic States (L genome: W=90, P=0.004).

Genetic diversity, HE, per pond in (a) the L genome and (b) the R genome. The x axis reflects increasing land area ranks based on land size and connectivity; however, within the ‘continent’, all four categories have the same land area rank (see Materials and methods).
In all-hybrid ponds, HE was highly significantly positively correlated between the L and R genomes, indicating that although they are propagated partially independently, the two genomes are affected similarly by processes like founder effect and drift (Spearman rank two-tailed test on all-hybrid ponds, rs=0.534, n=75, P=8.06e−07).
As expected, both HEL and HER correlated significantly and negatively with latitude (north) within all-hybrid ponds (n=75, Spearman rank two-tailed tests, HEL: rs=−0.557, P=2.10e−07 and HER: rs=−0.384, P=0.0007). This result was not confounded with the effect of land area, as latitude and the land area rank were not correlated (rs=−0.007, P=0.953). In the Baltic ponds, HEL showed a steep negative relationship with latitude, but this was not significant due to the low sample size (rs=−0.643, n=7, P=0.139).
Correlations between HE and longitude (east) were not significant for any of the genomes within all-hybrid ponds (n=75, HEL: rs=−0.098, P=0.404 and HER: rs=0.012, P=0.916). The correlation was almost significant for the R genome in the pooled Danish+Swedish ponds (n=66, rs=0.219, P=0.078), but not in Germany Inland ponds (n=13, rs=0.060, P=0.842).
Contrary to expectations, there were no significant positive correlations between genetic diversity and land area in all-hybrid populations (n=75, HEL: rs=0.120, P=0.307; HER: rs=0.160, P=0.171). Within Danish+Swedish ponds pooled, there was even a significantly negative correlation for the L genome (n=66, rs=−0.295, P=0.016). It thus appears that small and especially medium-sized Danish islands have water frog populations with high genetic diversity compared with large islands and, in particular, the Danish peninsula Jutland (Figure 2).
FST and isolation by distance
Global FST was 0.455 for the L genome and 0.580 for the R genome and similar to the values for the subset of all-hybrid ponds (L: 0.451 and R: 0.598).
In tests for isolation by distance, DS increased significantly with ln(km) in both genomes (Mantel tests on all ponds, L: r=0.502, P=0.00001; R: r=0.363, P=0.00001; Figure 3). Partial Mantel tests including the sea matrix were also highly significant for DS (L: r=0.461, P=0.00001; R: r=0.280, P=0.00001). However, whereas 34.2% of the total variance in the L genome was explained by ln(km), only 0.3% was additionally explained by the sea matrix. For the R genome, the corresponding values were 16.0 and 3.3%. Explanations for the low explanatory power of the sea matrix include high correlation between the geographic distance and the presence of sea between ponds (Mantel test, r=0.403, P=0.00001), and that only 19% of pond pairs were connected by land.

Isolation by distance in (a) the L genome and (b) the R genome. Colours represent connectivity between the pairs of ponds.
FST/(1−FST) gave less significant isolation by distance results than Ds; for the R-genome isolation by distance was not significant (Mantel tests: L: r=0.189, P=0.002; R: r=−0.006, P=0.448. Partial Mantel tests with sea matrix: L: r=0.151, P=0.019; R: r=−0.034, P=0.257). The proportions of the variance explained by the geographic distance, and, additionally, by the sea matrix were only 3.7 and 0.5%, respectively, for the L genome and 0.2 and 0.7% for the R genome.
Genomotype proportions in all-hybrid populations
On average, all-hybrid populations had 57.5% LLR, 33.9% LR and 8.2% LRR among males, and 19.3% LLR, 58.7% LR and 33.1% LRR among females. In addition, 0.38% LL was found among males and 0.08% among females. The proportions of the hybrid genomotypes showed large variation among ponds (Figure 4), forming a continuum rather than distinct population types.

Proportions of LLR, LR and LRR within males and females in Danish, Swedish and German ponds with all-hybrid populations of P. esculentus.
Male and female genomotype distributions were very different, mainly because LRR were rare in males but common in females throughout the distribution area of the all-hybrid populations (Figure 4). The few ponds with high LRR proportions in males were geographically scattered.
LLR female proportions decreased significantly toward the north and LLR and LR male proportions increased southeast and northwest, respectively (Table 2). Otherwise, there were no significant relationships between genomotype proportions and latitude, longitude or their interaction.
Pairwise differences in genomotype proportions between ponds increased significantly with geographic distance for males (Mantel test, n=77 ponds, r=0.075, P=0.035), but not for females (n=69 ponds, r=−0.015, P=0.378). Figure 5 suggests that pairwise differences in genomotype proportions increased with geographic distance for both sexes up to ∼3 ln(km) ∼20 km, beyond which they could not increase further.

Pairwise differences in genomotype composition of Danish, Swedish and German all-hybrid populations as a function of geographic distance for (a) males and (b) females. Shaded area indicates the rough distribution of data points. Ponds with <10 individuals per sex were excluded. The male data set contained 77 ponds and the female data set contained 69 ponds.
Genomotype versus habitat and body size
PCA of 15 habitat parameters gave six PCs (Table 3). Of these, PC2 (oxygen content, oxygen saturation, water temperature) was significantly negatively related to LR female proportions and significantly positively related to LRR proportions of both sexes (Table 4). PC4 (emerging vegetation, surface area, agriculture) was highly significantly negatively related to LRR female proportions, and PC5 (short grass, long grass) was significantly negatively related to LLR female proportions.
In German ponds, LLR female proportions increased significantly with forest on a scale from one to three (logistic regression, df=29, F=5.899, P=0.022) and LRR proportions of both sexes decreased significantly with forest (males: df=31, F=5.456, P=0.026; females: df=29, F=5.753, P=0.023). When including Swedish samples (12 ponds but more individuals per pond), only the decreasing relationship between LRR male proportions and forest remained significant (df=43, F=6.621, P=0.014).
Mean snout-to-vent lengths in all-hybrid ponds increased in the order LLR<LR<LRR, with males generally being smaller than females (mLLR: 65.5±5.0, mLR: 66.2±6.0, mLRR 73.9±8.2, fLLR: 70.5±7.7, fLR: 73.1±8.8 and fLRR: 75.0±9.7; Spearman rank two-tailed tests, males: rs=0.186, n=1237, P=4.11e−11; females: rs=0.160, n=1249, P=1.42e−08). However, LRR males were larger than both LLR and LR females. As size differences are related to ecological differences in mixed populations of diploid hybrids (LR) and parental species (LL, RR), the increase in size from LLR to LRR also suggests ecological differences between the genomotypes, with most differentiation between LLR and LRR.
Discussion
Genetic diversity and fragmentation
Our study on all-hybrid populations of the edible frog confirms the expectation that genetic diversity declines towards the northern distribution limit. This was expected for two reasons that cannot be disentangled: both post-glacial sequential founder events and the central-marginal model predict that higher isolation and lower effective population size decrease genetic diversity at a species’ range (Eckert et al., 2008). Nevertheless, the mean expected heterozygosity (HE) values of 0.157 for the L genome and 0.186 for the R genome found in all-hybrid populations are remarkably low. Although HE values depend on the markers used, so that comparisons between studies and species are problematic, our values appear exceptionally low compared with those from other amphibian studies at similar or higher latitudes (HE=0.319–0.614 in Andersen et al., 2004; Palo et al., 2004; Allentoft et al., 2009; Knopp and Merila, 2009). Only the very northern, small and isolated Norwegian and Swedish P. lessonae populations (Figure 1) are reported to have lower HE values (HE=0, Zeisset and Beebee, 2001). Further south, both P. lessonae and P. ridibundus had higher HE values demonstrating that the parental species are not always genetically depleted (HE=0.424–0.588, Zeisset and Beebee, 2001, 2003; Holsbeek et al., 2009). Our study confirmed that all-hybrid populations have significantly lower HE than adjacent populations to the south (Germany) and east (Baltic States).
A negative correlation between genetic diversity and land area has been shown in general (Frankham, 1996) as well as for moor frogs in almost the same study area as ours (Knopp et al., 2007). In this study, large islands, and especially the peninsula of Jutland, had unexpectedly low levels of genetic diversity compared with small and especially medium-sized islands. As low diversity at neutral markers does not necessarily imply low variability at coding genes under selection (Reed and Frankham, 2001), it cannot be predicted whether these populations will suffer from their low genetic diversity. However, it can be concluded that populations on even small islands are not particularly threatened by drift, as they are not more genetically depleted than populations elsewhere. The less intensive agriculture, and hence better habitat quality, of small islands might to some extent compensate for their small size by allowing large frog populations.
Our study found extremely high levels of population differentiation (FST=0.455 for the L genome and 0.580 for the R genome). Explanations include the large geographic area relative to the dispersal abilities of the frogs and the low microsatellite diversity. As differentiation cannot exceed the level of expected homozygosity within subpopulations, high levels of homozygosity allows for higher levels of differentiation (Hedrick, 1999).
Despite higher marker polymorphism in the R than L genome, isolation by distance was highest in the R genome. This could have unknown marker-related causes, or biological causes. One possibility is that genomotypes producing L gametes (P. lessonae and LLR) disperse more readily than those producing R gametes (LR, LRR and P. ridibundus). So far, no studies have to our knowledge investigated weather LLR, LR and LRR differ in their migration rates.
DS showed much more isolation by distance than FST. This difference is possibly because DS takes into account mutations, which are likely of importance given the several thousand years since colonization and fragmentation of the area. With DS, isolation by distance was found across the entire area sampled, although 81% of the ponds were separated by sea. Two explanations are possible for this apparently small sea influence. (1) The brackish sea water is not an important barrier to P. esculentus dispersal. In support of this, fishermen in the inner Danish waters sometimes catch P. esculentus when they set fish traps close to land (Fog et al., 1997). It is also possible that people occasionally transport eggs, tadpoles or frogs between islands for hobby purposes. (2) The populations in the area have not yet reached migration-drift equilibrium after fragmentation of the land area started about 7000 years ago. At least on the large islands and the continent, frog populations are likely to be large, and in large populations drift is small so that equilibrium is only very slowly approached. Two earlier studies on frogs in archipelagos show diverging results. One study found high levels of gene flow in the common toad and the common frog in a Finnish archipelago where the water has even lower salinity than this study (Seppa and Laurila, 1999). The second study found strong differentiation on Panamanian islands in the strawberry frog, that is in a terrestrial frog in an oceanic archipelago (Rudh et al., 2007).
All-hybrid distribution
In this study, all-hybrid populations were found as far south as ponds Sc and Sh (Figure 1). Thus, they extend beyond latitudes where another study found high levels of P. lessonae (near Serrahn between Da and Gr, Berger and Günther, 1988). A third study found high numbers of P. ridibundus south of ponds Sh and east of Ha and Her, supporting the conclusion from this study that the all-hybrid area ends this far south (near Steckby, Berger and Günther, 1991–1992).
Three things complicate the determination of the distribution of all-hybrid populations. First, the body length used for defining adults is critical, because the frequency of non-hybrids decreases steeply with size/age. Although adult males can be identified by the presence of vocal sac openings, adult females not carrying eggs differ from juveniles of both sexes only by their size. Second, detection of rare non-hybrids is strongly dependent on sample size. Third, survival of non-hybrids can depend on weather conditions, and may therefore vary temporally (Vorburger, 2001b). Hence, without established standard methods, the decision to call a population all-hybrid depends entirely on individual authors.
All-hybrid genomotype proportion differences
A continuous distribution of genomotype proportions was found, so that no clearly differentiated categories of population type were identified. Genomotype proportions were very variable on a small geographic scale, leaving less variation to be explained by geographic variation on a regional scale.
Instead, we found some evidence for an effect of habitat on genomotype proportions, in accordance with the phenotypic effect of genome dosage observed on body length (LLR<LR<LRR within both sexes). Consistent for both sexes, LRR proportions increased with PC2, representing oxygen content and saturation and, with less weight, water temperature. This corresponds well with literature statements that oxygen is an important and critical parameter for RR that usually live and breed in large, well-oxygenated water bodies and hibernate aquatically (reviewed by Plötner, 2005). As both LR and LRR females were frequent, their proportions were negatively correlated with one another, explaining the significant decrease of LR female proportions with PC2. In contrast, LRR males were so rare that their significant increase with oxygen would not necessarily cause significant decreases in LLR or LR proportions. Nevertheless, LLR males showed a strong negative trend with PC2. Although no explanations are at present available for the correlations of certain genomotypes with vegetation parameters (PC4, PC5 and wood), our results add to existing data suggesting that the habitat affects the proportions of LLR, LR and LRR (c.f. Peter et al., 2002; Jakob, 2007, Chapter 3).
How to remain all-hybrid
The low genetic diversity within the L and the R genome in all-hybrid P. esculentus, compared with adjacent populations and other amphibians (see above), seems to be compensated by high genetic heterozygosity within individual hybrid frogs. As almost all alleles were genome specific, every individual and pond had an overall expected heterozygosity of one, preventing inbreeding even in very small and homogeneous populations. Moreover, even if little genetic diversity is available for selection, the same genetic material can give rise to three different phenotypes due to dosage effect in di- and triploid individuals (LLR, LR and LRR). Although most genomotypes are required for reproduction, genomotype proportions and not just gene frequencies can quickly become adapted by natural selection.
Not only is low genetic diversity tolerable for the all-hybrid populations, it also seems to be a precondition for the inviability of non-hybrids and thus for the existence of all-hybrid populations in Denmark, southern Sweden and northern Germany. In other parts of Europe, single ponds with all-hybrid frog populations (listed in Christiansen et al., 2005) might be explained by ecological conditions being optimal for P. esculentus and suboptimal for the parental species. But our study area encompasses a range of habitats, some of which should be suitable for genetically healthy P. lessonae and/or P. ridibundus.
The diversity of breeding systems in the P. esculentus complex is unique among hybridogenetic taxa (four fishes, one stick insect and two other water frog complexes involving P. ridibundus; Christiansen and Reyer, 2009; Holsbeek and Jooris, 2009). This study shows that the all-hybrid system also contains a great variety of genomotype proportions. Thus, P. esculentus seems to be a natural experiment with potentially exciting outcomes if given time and space to proceed. Swedish all-hybrid populations are currently expanding their range (Fog et al., 1997; Arioli, 2007, Chapter 4). In Denmark, we have the impression from county offices and field herpetologists that the distribution area has shrunk since the account of Fog et al. (1997), but that P. esculentus is thriving in areas where ponds have been dug to promote endangered amphibian species.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008). Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17: 5177–5188.
Allentoft ME, Siegismund HR, Briggs L, Andersen LW (2009). Microsatellite analysis of the natterjack toad (Bufo calamita) in Denmark: populations are islands in a fragmented landscape. Conserv Genet 10: 15–28.
Andersen LW, Fog K, Damgaard C (2004). Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). P Roy Soc Lond B Biol 271: 1293–1302.
Arioli M (2007). Reproductive patterns and population genetics in pure hybridogenetic water frog populations of Rana esculenta. PhD thesis, University of Zurich, Zurich, Switzerland, http://www.dissertationen.uzh.ch.
Beebee TJC (2005). Conservation genetics of amphibians. Heredity 95: 423–427.
Berger L, Günther R (1988). Genetic composition and reproduction of water frog populations (Rana kl. esculenta Synklepton) near nature reserve Serrahn, GDR. Arch Natschutz Landschforsch Berlin 28: 265–280.
Berger L, Günther R (1991–1992). Inheritance patterns of water frog males from the environments of nature reserve Steckby, Germany. Zool Pol 37: 87–100.
Bogart JP, Bi K, Fu JZ, Noble DWA, Niedzwiecki J (2007). Unisexual salamanders (genus Ambystoma) present a new reproductive mode for eukaryotes. Genome 50: 119–136.
Bonnet E, Van de Peer Y (2002). zt: a software tool for simple and partial Mantel tests. J Stat Softw 7: 1–12 http://www.psb.ugent.be/∼erbon/mantel.
Chen BY, Wang MY, Lu WB, Chang JS (2007). Use of active consortia of constructed ternary bacterial cultures via mixture design for azo-dye decolorization enhancement. J Hazard Mater 145: 404–409.
Christiansen DG (2009). Gamete types, sex determination and stable equilibria of all-hybrid populations of di- and triploid water frogs (Pelophylax esculentus). BMC Evol Biol 9: 135.
Christiansen DG, Fog K, Pedersen BV, Boomsma JJ (2005). Reproduction and hybrid load in all-hybrid populations of Rana esculenta water frogs in Denmark. Evolution 59: 1348–1361.
Christiansen DG, Reyer HU (2009). From clonal to sexual hybrids: genetic recombination via triploids in all-hybrid populations of water frogs. Evolution 63: 1754–1768.
Eckert CG, Samis KE, Lougheed SC (2008). Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17: 1170–1188.
Fog K, Schmedes A, Rosenørn de Lasson D (1997). Nordens padder og krybdyr. Gad: Copenhagen, Denmark.
Frankham R (1996). Relationship of genetic variation to population size in wildlife. Conserv Biol 10: 1500–1508.
Frost DR, Grant T, Faivovich J, Bain RH, Haas A, Haddad CFB et al. (2006). The amphibian tree of life. Bull Am Mus Nat Hist 297: 8–370.
Graf JD, Polls Pelaz M (1989). Evolutionary genetics of the Rana esculenta complex. In: Dawley RM, Bogart JP (eds). Evolution and Ecology of Unisexual Vertebrates. New York State Museum Bulletin 466 New York State Museum: Albany, NY. pp 289–302.
Hammer Ö, Harper DAT, Ryan PD (2001). PAST: palaeontological statistics software package for education and data analysis. Palaeontol Electronica 4: 9 http://folk.uio.no/ohammer/past.
Hardy OJ, Vekemans X (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620 http://ebe.ulb.ac.be/ebe/Software.html.
Hedrick PW (1999). Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53: 313–318.
Holsbeek G, Jooris R (2009). Potential impact of genome exclusion by alien species in the hybridogenetic water frogs (Pelophylax esculentus complex). Biol Invasions (doi 10.1007/s10530-10009-19427-10532).
Holsbeek G, Maes GE, De Meester L, Volckaert FAM (2009). Conservation of the introgressed European water frog complex using molecular tools. Mol Ecol 18: 1071–1087.
Jakob C (2007). Structure and dynamics of pure hybridogenetic water frog populations of Rana esculenta in Southern Sweden. PhD thesis, University of Zurich, Zurich, Switzerland, http://www.dissertationen.uzh.ch.
Keller LF, Waller DM (2002). Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241.
Kier G, Kreft H, Lee TM, Jetz W, Ibisch PL, Nowicki C et al. (2009). A global assessment of endemism and species richness across island and mainland regions. Proc Natl Acad Sci USA 106: 9322–9327.
Knopp T, Cano JM, Crochet PA, Merila J (2007). Contrasting levels of variation in neutral and quantitative genetic loci on island populations of moor frogs (Rana arvalis). Conserv Genet 8: 45–56.
Knopp T, Merila J (2009). The postglacial recolonization of Northern Europe by Rana arvalis as revealed by microsatellite and mitochondrial DNA analyses. Heredity 102: 174–181.
Kort & Matrikelstyrelsen (2002). Det Levende Danmarkskort. Copenhagen, Denmark.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Palo JU, Schmeller DS, Laurila A, Primmer CR, Kuzmin SL, Merila J (2004). High degree of population subdivision in a widespread amphibian. Mol Ecol 13: 2631–2644.
Peter AKH, Reyer HU, Abt Tietje G (2002). Species and sex ratio differences in mixed populations of hybridogenetic water frogs: the influence of pond features. Ecoscience 9: 1–11.
Plötner J (2005). Die Westpaläarktischen Wasserfrösche - von Märtyrern der Wissenschaft zur Biologischen Sensation. Laurenti-Verlag: Bielefeld.
R Development Core Team (2009). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, http://www.R-project.org.
Reed DH, Frankham R (2001). How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55: 1095–1103.
Rudh A, Rogell B, Hoglund J (2007). Non-gradual variation in colour morphs of the strawberry poison frog Dendrobates pumilio: genetic and geographical isolation suggest a role for selection in maintaining polymorphism. Mol Ecol 16: 4284–4294.
Rybacki M, Berger L (2001). Types of water frog populations (Rana esculenta complex) in Poland. Mitt Mus Naturkd Berl Zool Reihe 77: 51–57.
Seppa P, Laurila A (1999). Genetic structure of island populations of the anurans Rana temporaria and Bufo bufo. Heredity 82: 309–317.
Som C, Reyer HU (2006). Demography and evolution of pure hybridogenetic frog (Rana esculenta) populations. Evol Ecol Res 8: 1235–1248.
Vorburger C (2001a). Fixation of deleterious mutations in clonal lineages: evidence from hybridogenetic frogs. Evolution 55: 2319–2332.
Vorburger C (2001b). Non-hybrid offspring from matings between hemiclonal hybrid waterfrogs suggest occasional recombination between clonal genomes. Ecol Lett 4: 628–636.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population-structure. Evolution 38: 1358–1370.
Wright S (1931). Evolution in Mendelian populations. Genetics 16: 97–159.
Zeisset I, Beebee TJC (2001). Determination of biogeographical range: an application of molecular phylogeography to the European pool frog Rana lessonae. P Roy Soc Lond B Biol 268: 933–938.
Zeisset I, Beebee TJC (2003). Population genetics of a successful invader: the marsh frog Rana ridibunda in Britain. Mol Ecol 12: 639–646.
Acknowledgements
We are grateful to Lars Iversen, Eline Embrects, Kåre Fog and Egidijus Sireika for help with sampling. We also thank many Danish, Swedish, German and Baltic colleagues for recommending sampling localities and many authorities and land owners for permission to sample frogs. We thank Sandra Röthlisberger for extraction and PCR, and Christian Jakob and Martina Arioli for providing genomotypes for some of the frogs. Lukas Keller, Peter Wandeler, Christoph Vorburger and Josh Van Buskirk kindly gave statistical advice, Christian Mayer was a good discussion partner and Josh Van Buskirk improved the manuscript. The study was supported by the Swiss National Fund (grant no. 31–64004.00 to H-U Reyer).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Christiansen, D., Reyer, HU. Effects of geographic distance, sea barriers and habitat on the genetic structure and diversity of all-hybrid water frog populations. Heredity 106, 25–36 (2011). https://doi.org/10.1038/hdy.2010.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.37
Keywords
This article is cited by
-
The role of deleterious mutations in the stability of hybridogenetic water frog complexes
BMC Evolutionary Biology (2014)
-
Genomic effects on advertisement call structure in diploid and triploid hybrid waterfrogs (Anura, Pelophylax esculentus)
BMC Ecology (2013)
-
Genetic structure of the marsh frog (Pelophylax ridibundus) populations in urban landscape
European Journal of Wildlife Research (2012)
-
Carlquist revisited: history, success, and applicability of a natural history model
Biology & Philosophy (2012)
