
Abstract
Understanding the natural history of model organisms is important for the effective use of their genomic resourses. Arabidopsis lyrata has emerged as a useful plant for studying ecological and evolutionary genetics, based on its extensive natural variation, sequenced genome and close relationship to A. thaliana. We studied genetic diversity across the entire range of European Arabidopsis lyrata ssp. petraea, in order to explore how population history has influenced population structure. We sampled multiple populations from each region, using nuclear and chloroplast genome markers, and combined population genetic and phylogeographic approaches. Within-population diversity is substantial for nuclear allozyme markers (mean P=0.610, Ae=1.580, He=0.277) and significantly partitioned among populations (FST=0.271). The Northern populations have modestly increased inbreeding (FIS=0.163 verses FIS=0.093), but retain comparable diversity to central European populations. Bottlenecks are common among central and northern Europe populations, indicating recent demographic history as a dominant factor in structuring the European diversity. Although the genetic structure was detected at all geographic scales, two clear differentiated units covering northern and central European areas (FCT =0.155) were identified by Bayesian analysis and supported by regional pairwise FCT calculations. A highly similar geographic pattern was observed from the distribution of chloroplast haplotypes, with the dominant northern haplotypes absent from central Europe. We conclude A. l. petraea's cold-tolerance and preference for disturbed habitats enabled glacial survival between the alpine and Nordic glaciers in central Europe and an additional cryptic refugium. While German populations are probable peri-glacial leftovers, Eastern Austrian populations have diversity patterns possibly compatible with longer-term survival.
Similar content being viewed by others

Introduction
Understanding the natural history of model organisms is important for the effective use of their genetic resources, aiding sampling strategies as well as data analysis and interpretation. Neutral variation is often structured in natural populations by historical and demographic processes (Hewitt, 2004). Understanding what role these processes have had is essential when attempting to identify polymorphisms that are structured by adaptation and selection processes (Nielsen, 2005).
Arabidopsis lyrata (L.) (Al-Shehbaz and O’Kane, 2002) has emerged as a useful organism for plant ecological and evolutionary studies, benefiting from the wealth of molecular resources from its close congenitor Arabidopsis thaliana (L.) Hyenh. (Koch et al., 2000; Mitchell-Olds, 2006), and now from its own fully sequenced genome. Arabidopsis lyrata is an outcrossing perennial that shows extensive phenotypic variation and grows in natural habitats that span a large ecological range (Jonsell et al., 1995; Kivimäki et al., 2007; Leinonen et al., 2009; Sandring and Ågren, 2009). Already several local adaptation/selection studies have been published (Kivimäki et al., 2007; Kuittinen et al., 2007; Leinonen et al., 2009), with special emphasis on studying self-incompatibility evolution (Charlesworth et al., 2003; Schierup et al., 2006; Mable and Adam, 2007). More of such studies are likely to emerge with the recent availability of the genomic sequence, which makes a comprehensive study of A. lyrata's phylogeographic structure timely.
The A. lyrata complex has a highly disjunct circumpolar distribution range. We follow the taxonomic treatment of Al-Shehbaz and O’Kane, (2002) and here focus on the European taxon A. lyrata subsp. petraea (hereafter Arabidopsis l. petraea). This taxon has been the most extensively studied member of the group, due to its well-documented distribution and collecting accessibility (Jalas and Suominen, 1994). Within Europe, A. l. petraea has a highly disjunct distribution, because of an ecological preference for highly disturbed low-competition habitats (Ratcliffe, 1994). In central Europe it is confined to discrete areas within Hungary, eastern Austria, Czech Republic, southern and central Germany, whereas in northern Europe it has a wider range, from Iceland, central Norway, the Swedish coast and the British Isles. With such a fragmented distribution range, population history is likely to have an important role in shaping genetic structure.
Quaternary glacial cycles over the last 2.5 million years have greatly affected the genetic structure of European plants and animals (Hewitt, 2004). During the last glacial maximum (c. 18 000 years bp), massive glaciers formed across northern Europe and locally in the Alps and Pyrenees, and intervening landscape developed a tundra environment. Long-term species survival is typically dependent on population persistence in ice-free locations (refugia) south of the glacier and tundra limits, resulting in geographic isolation and genetic differentiation (Petit et al., 2003; Hewitt, 2004). Rapid population expansion in the post-glacial period causes sequential bottlenecks (Ibrahim et al., 1996) and thus further altering population structure. However, comparative analysis indicates species have highly individualistic responses to climate change (Taberlet et al., 1998), varying according to refugia locations, ecological preferences, reproductive biology and dispersal mechanisms. For cold-tolerant taxa, alternative northern and mountain survival histories are reported (Alsos et al., 2005; Ehrich et al., 2007; Mráz et al., 2007), further complicating Europe's phylogeographic history.
Over the last decade regional and larger scale studies have provided insights into the genetic structure of A. l. petraea populations (Jonsell et al., 1995; van Treuren et al., 1997; Clauss and Mitchell-Olds, 2006; Gaudeul et al., 2007; Muller et al., 2008). A phylogeographic model of central European survival, with subsequent post-glacial population expansion into northern Europe was proposed after analysing microsatellite variation (Clauss and Mitchell-Olds, 2006; Muller et al., 2008). Similar conclusions were reached by a recent SNP analysis (Ross-Ibarra et al., 2008). However, existing studies are based on low sampling densities relative to the local abundance and fragmented distribution range. Regional studies with locally dense sampling indicate that Icelandic and Scandinavian samples maintain extensive intra-population diversity (Jonsell et al., 1995; Gaudeul et al., 2007), and that Scandinavian populations may have not been subject to recent genetic bottlenecks (Gaudeul et al., 2007). Furthermore, Kivimäki et al., (2007) showed that northern populations have allelic diversity at the FRIDGA locus that is missing from central European populations. These observations do not readily conform to the expectations of rapid post-glacial population expansion from a central European refugium.
A detailed genetic study based on a comprehensive sampling of the entire European range is missing. Existing studies show that population differentiation is relatively high over short geographic distances (Schierup, 1998; Clauss and Mitchell-Olds, 2006), and diversity is locally substructured (Gaudeul et al., 2007), and therefore uniform and dense sampling would assist in the detection of wider geographic structure. We favour a combination of phylogeographic and population genetics approaches. The latter should ideally involve a codominant marker system, allowing for detection of bottlenecks and within-population inbreeding, which are predicted to alter during colonization processes (Hewitt, 2004). Codominant markers also permit the discrimination between diploid and polyploid populations, which distributions are presently uncertain in central Europe. Allozymes are suited for these purposes, allowing for a rapid screen of many populations and a detailed geographic sampling, and have already been applied to earlier studies (Jonsell et al., 1995; van Treuren et al., 1997; Kärkkäinen et al., 1999).
In this study, we aim to (1) determine the number and distribution of discrete genetic clusters; (2) to investigate how within-population diversity, inbreeding and genetic bottlenecks are spatially organized; (3) to determine if northern populations are a subset of central European diversity and (4) examine the proposed phylogeographic hypothesis of northern recolonization from a central European source. Finally, we discuss the wider implications of our study for future A. lyrata model organism research.
Materials and methods
Sampling and polyploidy determination
Leaf material was collected from 1724 individuals of A. l. petraea from 59 populations in Hungary, Austria, Czech Republic, Germany, UK, Ireland, Iceland, Norway and Sweden (Supplementary Table S1 and S2), thereby covering nearly the entire European distribution range (Jalas and Suominen, 1994). Plants were collected at least 1 m apart. Both diploid (2n=16) and polyploid (2n=32) chromosome counts are reported for populations in Austria and the Czech Republic (Polatschek, 1966; Hejeny and Slavik, 1992), and we interpreted departures from diallelic allozyme staining patterns as evidence for polyploidy. Polyploids were identified in 17 populations (Supplementary Table S2 and Supplementary Figure S1), and genetic data analysis was conducted on the remaining 42 diploid populations (Supplementary Table S1). These originate from: Austria (n=7), Germany (n=9, Bavaria and Harz Mountains), Czech Republic (n=1), the British Isles (n=12, Republic of Ireland, Wales, Scotland, Shetland), Iceland (n=6), Norway (n=5) and Sweden (n=2). Voucher specimens for the majority of population were deposited at the Natural History Museum (London) molecular collections store.
Allozyme variation
Fresh leaf material was ground in a Tris-HCl (pH 7.5) extraction buffer and proteins were fractionated on 12.5% hydrolysed potato starch gels under standard procedures. The following enzyme systems were resolved according to Ansell et al., (2008): AAT (E.C. 2.6.1.1), PGI (E.C. 5. 3. 1. 9), PGM (E.C. 5. 4. 2. 2), TPI (E.C. 5. 3. 1. 1), ADH (E.C. 1.1.1.1), IDH (E.C. 1.1.142), 6-PGD (E.C. 1. 1. 1. 44), SKDH (E.C. 1. 1. 1. 25), UGPP (E.C. 2. 7.9).
Arabidopsis. l. petraea is capable of vegetative reproduction and clones (identical multi-locus genotypes, MLGs) were identified in 28 of the 42 diploid populations when the genotypes were sorted in Microsoft EXCEL. These accounting for 1.16–36.67% of within-population sampling (Supplementary Table S1), and after excluding these, genetic analysis was performed on the remaining 1203 individuals. To test for independence of loci, linkage disequilibrium was calculated among all pairs of polymorphic loci in each population using GDA v.1.16 (Lewis and Zaykin, 2001), applying a liberal sequential Bonferroni correction of 10 times threshold (that is, P=0.005) for a conservative assessment of loci independence. To detect recent bottlenecks, deviations from a mutation-drift equilibrium were examined by a one-tailed Wilcoxon sign-ranked test for an excess of heterozygosity (Cornuet and Luikart, 1996), following the infinite allele model (IAM), using the BOTTLENECK program v. 1.202 (Piry et al., 1999). The following measures of genetic variability were calculated on a per population basis: proportion of polymorphic loci (P) (a locus was considered polymorphic when the most common allele did not exceed a frequency of 0.95), the effective number of alleles per locus (Ae), expected heterozygosity (He), observed heterozygosity (Ho), and inbreeding coefficient (f) using POPGENE v.1.31 (Yeh et al., 1999). Geographic patterns in diversity were assessed by two-tailed Mann–Whitney U-tests, and linear regressions of diversity against decimalised degrees (Stern et al., 2005), using INSTAT v. 3.036 (University of Reading, UK).
Population structure and inbreeding was assessed by the analysis of variance procedure (Weir and Cockerham, 1984), which provides estimators (f, F, θ) of F-statistics, FIS, FIT, FST. Hierarchical analyses of FST (θ) were conducted to assess the extent of differentiation between (FCT) and within (FSC) regions. Ninety-five percent confidence intervals were calculated for all F-statistics by bootstrap re-sampling (9999 replicas) and GDA. The relationship between genetic similarity (log FST) and geographic distance (logKm) was investigated by Mantel tests via the R-package v. 4.00 (Casgrain and Legendre, 2001). Geographic distances were derived from the longitude and latitude co-ordinates (Stern et al., 2005) using the R-package. Population structure was further investigated by a Bayesian-based assignment algorithm (incorporating geographic information), as implemented by the STRUCTURE program v.2.3.1 (Pritchard et al., 2000). Analyses followed the admixed model with independent allele frequencies, employing 504 MCMC replicates, including 104 for burn-in period, with 10 simulations per cluster (K), and the minimum number of K was evaluated using the delta-K procedure (Evanno et al., 2005)
Chloroplast sequence variation
Chloroplast DNA sequence variation in the non-coding intergenic spacer (IGS) region between the trnL(UAA) and trnF(GAA) genes (trnL-F IGS) was examined in 10 individuals per diploid population (except Luglas with n=20). In Arabidopsis, this region contains a variable number of partially duplicated trnF gene (pseudogene) copies, with a complex pattern of evolution (Ansell et al., 2007; Schmickl et al., 2009). However, the region 5′ to the duplications follows a simple mutation pattern that is suitable for haplotype delimitation (alignment positions 1–180, Supplementary Figure S1; Ansell et al., 2007). Data for 37 of the 42 A. l. petraea populations was obtained from an earlier study (Ansell et al., 2007), and new sequences were generated for an additional five populations (Norway NO1, 2, 3, 4 and Republic of Ireland IR1). DNA extractions, PCR amplifications and DNA sequencing of the trnL-F IGS followed the conditions described in Ansell et al., (2007).
DNA sequences were manually aligned using BIOEDIT v. 7.05 (Hall, 1999), and two new trnL-F sequences were identified and submitted to GenBank (accession numbers GU456721-GU456722 for PET 35, 36). The partial trnF(GAA) gene duplications (trnF pseudogenes) were identified according to the boundaries definitions of Ansell et al., (2007), which differ from those of Schmickl et al., (2009). The fragment portion 5′ of the duplications was used to assign haplotypes to individual sequence. No new haplotypes were recovered compared with the related earlier publication of Ansell et al., (2007) and the haplotype network presented is reproduced from this study, with a minor modification, the network was rooted to the trnL-F IGS sequences of A. thaliana reported in Beck et al., (2008). Haplotype richness (R) with correction for the smallest regional sample size (n=60) through rarefaction was estimated using CONTRIB (Petit et al., 1998). Nucleotide diversity and genetic differentiation were calculated using ARLEQUIN v. 3.11 (Excoffier et al., 2005). Genetic differentiation was initially derived from haplotype frequencies (FST) and thereafter by incorporating the genetic distances (as the number of character differences) that separate all pairs of haplotype to estimate ΦST. The significance of differentiation (FST and ΦST) was assessed by 10 000 non-parametric permutation tests in ARLEQUIN. Demographic history was explored by mismatch distribution analyses of observed haplotype pairwise differences, following a sudden population expansion model (Rogers and Harpending, 1992), employing 100 bootstraps, using ARLEQUIN.
Results
Allozyme diversity
Genetic variation was examined in nine enzyme systems and 12 polymorphic loci were scored (Supplementary Table S3). In total 68 alleles were detected among the 42 diploid populations, and 15 alleles were confined to two or fewer populations. In addition, six alleles were confined to central Europe and a further seven were confined to northern Europe (Supplementary Table S3). Exact tests between pairs of polymorphic loci were performed separately for the central and northern European populations, reflecting the major population sub-division (see later). Five and six non-identical loci pairings from central and northern European data sets respectively, had significant linkage disequilibria after sequential Bonferroni correction for multiple tests (Table 1). Overall the loci were judged statistically independent. A large proportion of populations (23 of 42) had deviations (<P=0.05) from mutation-drift equilibrium under the one-tailed Wilcoxon's test for an excess of heterozygosity (Supplementary Table S1), indicating recent genetic bottlenecks. The proportion of bottlenecked populations was broadly similar among both central (8 of 17) and northern (15 of 25) European populations.
Considerable intra-population variation was detected (Table 2), as shown by mean within-population estimates of proportion polymorphic loci (P) of 0.610, effective number of alleles per locus (Ae) of 1.580 and expected heterozygosity (He) of 0.277. Mann–Whitney tests detected non-significant differences in P, Ae, He, Ho between the central and northern European populations (P=0.124–0.818). Comparing the main sampling areas within central Europe, the Austrian populations had significantly higher P (P=0.044), but non-significantly different Ae, He, Ho compared with southern Germany (P=0.064–0.247). Comparing the main sampling areas within northern Europe, all Mann–Whitney tests among the British Isles, Iceland and Scandinavia were non-significantly different (P=0.062–0.961), except Ae (P=0.035), He (P=0.052), Ho (P=0.011) were significantly lower in Scandinavia compared with the British Isles, and Ho was lower in Iceland than Scandinavia (P=0.011). Regional comparisons across central and northern Europe were non-significantly different (P=0.106–0.866), except P was higher in Austria compared will Scandinavia (P=0.042), and southern Germany had significantly higher Ae (P=0.037) and He (P=0.049) compared with Scandinavia. Linear regressions analysis of P, Ae and He against decimalized degrees north or east were non-significant (r2=0.001–0. 012, P=0.168–0.993). There was a small departure from Hardy–Weinberg equilibrium over the entire data set FIS=0.129 (95% CI 0.066, 0.185 Table 3), and inbreeding was higher among northern FIS=0.163 (95% CI 0.078, 0.243) compared with central European populations FIS=0.093 (95% CI 0.052, 0.134). A Mann–Whitney test revealed significantly higher within-population inbreeding coefficient (f) for northern populations compared with central European populations (P=0.013). A linear regression of f against decimalized degrees north was significant (r2=0.264, P=0.005), but non-significant against decimal degrees east (r2=0.021, P=0.360).
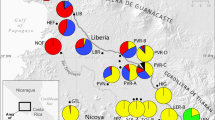
The number of population clusters (K) within the entire dataset was explored using Bayesian analysis. The log likelihood was calculated over K=1–6 with STRUCTURE, and the delta-K analysis indicates that K=2 is most likely the correct minimum number of clusters (Figure 1). K2 separates populations from central Europe and northern Europe (Figure 2a), with populations typically having 92–99% assignment to a single cluster, but with evidence of local recent genetic admixture for populations in Iceland and western Scotland (Supplementary Table S1). Population structure was further explored by conventional F-statistic analysis. A hierarchical FST analysis indicated that diversity is partitioned between the central and northern Europe populations FCT=0.155 (95% CI 0.067, 0.230). This trend was further observed when FST was then calculated among all pairwise combinations of regions with multiple population samplings (Figure 2b and Table 3). Differentiation was consistently lower for the regional comparisons from within central or northern Europe (FST ranging 0.042–0.088), compared with tests that involved both central and northern Europe areas (FST ranging 0.106–0.259). Populations from northern Europe had higher differentiation FST=0.249 (95% CI 0.202, 0.0294) compared with the central European populations FST=0.148 (95% CI 0.128, 0.169), and overall, European-wide differentiation was FST=0.271 (95% CI 0.212, 0.326). A Mantel test indicated a relationship (r2=0.225, P=0.001) between genetic similarity (log FST) and geographic distance (log Km) when calculated among all 861 pairwise combinations of populations.

Plot of mean posterior probability (LnP(D)) values (open circles) per clusters (K), based on 10 replicates per K, generated by the STRUCTURE program (Pritchard et al., 2000), and delta-K analysis (filled squares) of LnP(D), according to Evanno et al., (2005).

Geography structuring of genetic diversity among populations of A. l. petraea with allozyme staining patterns consistent with diploidy. Allozyme dataset: STRUCTURE program (Pritchard et al., 2000) assignment scores for K=2 clusters, with B letters denoting populations with genetic bottlenecks (a) and inter-regional pairwise FST estimates (b). Chloroplast trnL-F IGS haplotype data set: inter-regional pairwise ΦCT estimates, with values in brackets denoting estimates excluding populations IC1-4 (c), frequency and distribution of haplotypes and genealogical relationships, with black lines indicating single mutation steps, and data depicted in squares derived from Koch and Matschinger, 2007 (d). Distribution range of A. l. petraea (dark grey) adapted from Jalas and Suominen, (1994) with isolated populations denoted by crosses. See Supplementary Table S1 for sampling information.
Chloroplast diversity
Two new trnL-F types (PET 35–36) were identified from the alignment of the new Norwegian and Irish sequences (Supplementary Table S4). After excluding the trnF pseudogene portion of the fragment, all sequences had haplotypes identical to those reported earlier Ansell et al., (2007) (Table 4). The distribution and frequencies of the 10 unique haplotypes is shown in Table 2, and the haplotype network in Figure 2d. Only haplotypes 2 and 6 occurred at high frequencies, 47 and 41% respectively. The remaining haplotypes were private to two or fewer populations in northern Europe (haplotypes 3, 7–10) or central Europe (haplotypes 1, 4–5). Haplotype 6 was confined to northern Europe, whereas haplotype 2 was present in both central and northern Europe, the latter at much lower frequencies and only common on eastern Iceland.
Haplotype richness by rarefaction (R) and nucleotide diversity (π) was higher for pooled northern European samples R=5.606 π=0.0047, compared with central European samples R=3.000 π=0.0026. At a regional scale, haplotype diversity was highest in the British Isles R=4.197, followed by Austria R=3.000, Scandinavia R=1.857 and Iceland R=1.000. Only haplotype 2 is present in southern Germany. Nucleotide diversity in contrast was highest in Iceland π=0.0053, followed by Austria π=0.0052, the British Isles π=0.0038, Scandinavia π=0.0018. Chloroplast diversity was significantly partitioned among European populations (Table 3), when estimated from haplotype frequencies FST=0.840 (P=0.0001), or when genetic distances were incorporated ΦST= 0.851 (P=0.0001). Diversity was partitioned between central and northern Europe areas ΦCT=0.581 (P=0.0001). This pattern was largely supported when ΦCT is calculated among all pairs of regions (Figure 2c, Table 3). Iceland was the exception to this trend, with high frequencies of haplotype 2 in populations IC1-4 (Figures 2c and d), resulting in Iceland having low overall differentiation from Austria and southern Germany. The remaining Icelandic populations (IC5-6, Figure 2d) had high frequencies of haplotype 6, and low pairwise estimates of ΦCT were obtained between IC5-6 and the British Isles and Scandinavia, values in brackets of Figure 2c.
Mismatch analyses on observed haplotype differences failed to reject the sudden population expansion model (Figure 3), when performed separately on cluster I and cluster II (northern European haplotype 6-10) haplotype lineages, with P=0.540 and P=0.580, respectively (Figures 2a and 3b). Individual tests on the following regions also failed to reject the sudden expansion model: cluster I Austria lineages P=0.333 (Figure 2d), cluster II British lineages P=0.680 and cluster II Scandinavian lineages P=0.140. However, the sudden expansion model was rejected for the central Europe cluster I lineages (haplotypes 1, 2, 4, 5) P=0.002 (Figure 2c). This result was biased by the relatively high density of sampling in southern Germany (n=8, 20 km × 30 km area), where the populations are invariant for haplotype 2.

Mismatch analysis of observed haplotype differences. All cluster I haplotype data (a) all cluster II haplotype data (b), central European cluster I data only (c), Austrian cluster I haplotype data only (d). Expected results dotted line, observed results solid line, P-values for rejection of sudden expansion model (Rogers and Harpending, 1992) are indicated.
Discussion
Polyploid populations were detected in areas that had previously been subjected to extensive cytological screening (Austria and Czech Republic, Polataschek, 1966; Hejeny and Slavik, 1992), but also those lacking detailed cytological data (northern Germany, and Hungary, Supplementary Figure S1). Thus our study helps to define the range and extent of diploid A. l. petraea. Within diploid A. l. petraea most diversity (c. 70%) was maintained within populations, which is consistent with early predictions made for this obligate outcrossing (self-incompatible SI) species (Clauss and Mitchell-Olds, 2006; Schierup et al., 2006; Mable and Adam, 2007). The mean estimates for diversity measures were higher compared with average estimates for other short-lived outcrossing perennials (Hamrick and Godt, 1996), potentially reflecting the prolonged flowering period and insect pollination that promote local gene flow.
For an outcrossing plant genetic variation was unusually structured at all geographic scales, with significant differentiation detected at the continental, regional and local scales, for example, Europe FST=0.271, central Europe FST=0.148, Norway FST=0.315, southern Germany FST=0.154. Our continental estimate was lower than that reported by a recent microsatellite study (FST =0.420, Muller et al., 2008), but our regional estimates are consistent with earlier allozyme and microsatellite studies (FST =0.17 and FST =0.252 for central Europe and Sweden, respectively Clauss and Mitchell-Olds, 2006; van Treuren et al., 1997). Recent demographic history has contributed to this widespread population differentiation, with genetic bottlenecks detected from c. 50% of populations and these are, approximately, uniformly distributed throughout the range (Figure 2a). Austria was an exception. Here, only two somewhat unusual populations showed bottlenecks, AU5/Luckerter from an old quarry, and AU7/Bärenschützklamm, a single patch colony. Furthermore, our results from chloroplast diversity revealed a pattern consistent with recent sudden population expansion (Figure 3). Thus we consolidate previous observations (Muller et al., 2008; Ross-Ibarra et al., 2008) and conclude that nearly the entire range of A. l. petraea has been strongly influenced by recent historical events.
Our dense sampling regime helped to minimize the effect of the local genetic drift, and we detected that diversity is organized at the continental scale in two population clusters, one in central Europe and one in northern Europe (Figures 2a–d). This arrangement mirrors the core divisions in the European distribution pattern (Jalas and Suominen, 1994), and was clearly supported by Bayesian and FST analyses of allozyme variation, and by the analysis of chloroplast sequences (Figures 2a–d). We acknowledge that by excluding the trnF pseudogene copies (Ansell et al., 2007; Schmickl et al., 2009) the remaining portion of the chloroplast sequences are short and additional sequencing might improve the phylogeographic resolution. Nevertheless, our data clearly indicate a distinct geographic substructuring of Europe diversity.
Iceland is the notable exception to the clear central–northern European split, with eastern populations (IC1-4) having high frequencies of a haplotype more typical of central European samples (Figure 2d). In Iceland and in western Scotland we recovered clear evidence of gene pool amalgamation from the allozymes (Figure 2a), indicating local gene flow in otherwise distinct populations. Recent coalescence analysis on 77 nuclear loci obtained a divergence time of around 36 000 years ago between Swedish and German samples (Ross-Ibarra et al., 2008). This would suggest the two European populations differentiated before the last glacial maximum (LGM) around 18 000 years ago (Hewitt, 2004), and the local genetic admixture is post-glacial in origin.
Our results contrast with two earlier European-wide diversity studies, which did not recover a distinct northern European population (Muller et al., 2008; Ross-Ibarra et al., 2008). These studies elected to investigate wider genetic diversity patterns and included samples of North American A. l. lyrata (Muller et al., 2008; Ross-Ibarra et al., 2008). Multi-locus sequencing studies have established that the European (A. l. petraea) and North American (A. l. lyrata) taxa are substantially differentiated (Wright et al., 2003, 2006). Thus inter-subspecies differences may have masked the more subtle substructuring of European diversity. Muller et al., (2008) also reported relatively high levels of homoplasy in their data, and the use of relatively few European populations (n=6) may have exacerbated this.
Compared with the central European populations, the northern populations showed only modest and non-significant reductions in diversity (P, He, Ho) (Table 2). This trend was further observed when populations of less than 92% assignment to a single Bayesian cluster were excluded from Mann–Whitney tests (except Ho, P=0.011). This contrasts with the earlier reports of significant reductions reported in comparative analyses (Clauss and Mitchell-Olds, 2006; Muller et al., 2008), although these studies included only limited northern sampling. In agreement with Muller et al., (2008) we found elevated levels of inbreeding for northern Europe, but these levels remained low (FIS=0.093 versus 0.163). Inbreeding increased locally in Iceland and Scandinavia (Table 3), but also remained low in comparison with the North American A. l. lyrata, where selfing probably evolved in the recent post-glacial period (Mable and Adam, 2007). The European A. l. petraea retains sporophytic self-incompatibility (Schierup, 1998; Charlesworth et al., 2003), the genetic control ensuring that populations must be established by at least two unrelated individuals. Schierup et al., (2006) detected 11 SRK (pistile locus) haplotypes in an Icelandic population, implying a minimum of six diploid foundering individuals. Consequently the SI system may have helped to retain diversity, counterbalancing the effects of rapid population expansion (Ibrahim et al., 1996) and explain the absence of a clear ‘southern richness versus northern purity’ pattern associated with many organism's recent post-glacial history (Petit et al., 2003; Hewitt, 2004).
Phylogeographic model development
There is increasing fossil and molecular evidence supporting the glacial survival of cold tolerant taxa north of the Alps during the last glacial maximum (LGM) (Alsos et al., 2005; Mráz et al., 2007), where an extensive tundral landscape developed between the Alps and northern German ice sheets (Lang, 1994). A. l. petraea is tolerant of cold conditions, having been recorded from 1250 m altitude in Scotland (Ratcliffe, 1994), and has a preference for natural disturbed habitats (Ratcliffe, 1994; Hemp, 1996). Consequently A. l. petraea potentially had a widespread distribution across central Europe during the LGM. Hence, it is proposed that A. l. petraea survived the LGM in central European localities and then recolonized northern Europe in the post-glacial period (Clauss and Mitchell-Olds, 2006; Muller et al., 2008). These studies cite the German population's maintenance of extensive heterozygosity, mutation-drift equilibrium and low within-population inbreeding as factors associated with an established population history and glacial survival. Our denser survey of 17 central European populations detected similar genetic patterns and broadly supports this hypothesis, but also indicates a more complex phylogeographic history. In agreement with the Arabidopsis genus-wide phylogeographic trends (Koch and Matschinger, 2007), eastern Austria is the centre of central European genetic diversity for A. l. petraea. Although centres of diversity may also arise from population amalgamation (Petit et al., 2003), and both diploid and polyploid populations of A. l. petraea occur in Eastern Austria (Polatschek 1966 and Supplementary Figure S1), the delta-K analysis of STRUCTURE outputs did not support the local involvement of a second gene pool, such as related A. arenosa (Figures 1 and 2a). Furthermore, there is little evidence of recent colonization within Austria, with low local estimates of differentiation and inbreeding (FST=0.063, FIS=0.069). Hence, Austria appears to have functioned as a long-term glacial refugium for A. l. petraea. This is in agreement with the findings from other cold-tolerant plants (Ehrich et al., 2007; Mráz et al., 2007). Relative to eastern Austria bottlenecks are widespread in Southern Germany, allozyme diversity is reduced and differentiation is elevated (FST=0.154), and the populations lack chloroplast variation despite comprehensive sampling (n=260, Ansell et al., 2007). Furthermore, a positive Tajima's D was obtained from Plech, implying this population and potentially the entire southern Germany range is not in equilibrium (Ross-Ibarra et al., 2008). In addition, the local chloroplast haplotype and a high fraction of allozyme diversity can be recovered from Austrian populations. Taken together, these observations suggest southern Germany is a secondary centre for post-glacial survival, derived from earlier Quaternary expansions from Austria, but having now reached a near equilibrium state (Ross-ibarra et al., 2008).
The long-term glacial survival of A. l. petraea in central Europe is linked to special local micro-habitats. Plant community analysis revealed that Bavarian A. l. petraea populations occupy special azonal habitats in semi-shaded dolomitic rocks, which have ecologies that closely resemble the cool moist rocky conditions that prevailed locally during the LGM (Hemp, 1996). Thus a local continuity of ecologies is provided in both warm and cold Quaternary periods, maintaining local genetic diversity. Similar habitats occur in the Wienerwald forests of the eastern Austrian Alps, where A. l. petraea is locally common (Polatschek, 1966). However, with each glacial cycle only subsets of individuals from the large glacial population are likely to survive on the special micro-site habitats. Consequently, A. l. petraea may have a history of glacial survival through a series of transitory glacial relict sites, rather than through a persistent recognizable refugial population.
Our data do not support the hypothesis of northern post-glacial colonization from a central European source (Clauss and Mitchell-Olds, 2006; Muller et al., 2008; Ross-Ibarra et al., 2008). We did not detect a clear reduction in northern European nuclear genome diversity, and the dominant northern cpDNA haplotypes are absent from extant diploid central European populations. This asymmetric haplotype distribution could be explained by the chloroplast genome having a small effective population size, making it vulnerable to drift during Quaternary population oscillations (Petit et al., 2003). However, the divergence time between the central-northern European populations based on nuclear loci appears to substantially predate the LGM (Ross-Ibarra et al., 2008), suggesting the population subdivision is not very recent. A similar asymmetric allelic distribution pattern was also observed from a survey of diversity at the FRIGDA locus (Kuittinen et al., 2007), which showed the central European populations lack the common northern allele. We speculate the northern populations are derived from a source population independent of the central Europe stock (for example northern Europe or Siberia), or represent a fraction of diversity that is no longer extant in the central European range. We argue it is likely A. l. petraea was capable of surviving around the margins of the northern European ice-sheets, as proposed for a number of other arctic-alpine species (Gabrielsen et al., 1997; Alsos et al., 2005; Ehrich et al., 2007). In Iceland A. l. petraea is only absent from the areas permanently glaciated, and fossilized seedpods are reported from former glaciated area of northern England in the early post-glacial (Jones, unpublished), suggesting glacial populations were nearby. Taken together these observations indicate colonization may have proceeded rapidly from source areas near to glacier margins; and that colonization proceeded at the margin of the receding glaciers, then climate warming exposed open tundral habitats as suitable niches for A. l. petraea to grow. The exact location of any potential northern refugia still remains cryptic.
Model organism implications
The development of Arabidopsis lyrata for studying plant ecological and evolutionary processes (Mitchell-Olds, 2006) will continue to gather pace with the recent arrival of the completed genome (http://www.jgi.doe.gov/sequencing/why/3066.html). So far a large proportion of adaptation and diversity studies have used ‘standard’ sampling locations (for example Esja Mountain, Karhumaki, Mjällom, Plech, Reykjavik). A growing number of studies have used collections from both central and northern European territories (Kuittinen et al., 2007; Leinonen et al., 2009), which we show here to be genetically structured. Future adaptation studies should ideally continue this wider sampling approach when testing for local adaptation.
In general few sampling sites are needed to capture a high proportion of Europe's allelic diversity (neutral variation), as a good portion of alleles are common among all populations. Nevertheless, the occurrence of locally restricted alleles (Supplementary Table S3) should be considered when developing seed stock centres, as is standard for A. thaliana research. Sampling should also consider the extensive local habitat diversity; such as gypsum, serpentine and dolomitic substrates, and the sea level to 1250 m altitudinal range. Presently it appears adaptation studies have almost avoided sampling populations from Austria (Clauss and Mitchell-Olds, 2006), which we here show to be the centre of central European genetic diversity. This under representation may reflect sampling uncertainties created by the lack of clear geographic organization of ploidy levels (Supplementary Figure S1), but we hope our results will enable targeted collecting of appropriate populations in this important region.
Our study highlights that genetic bottlenecks, recent demographic history and regional glacial survival are dominant factors in structuring European diversity. Consequently genomic studies should consider population history when testing hypothesis, for example, selective sweeps and genome scans. While recombination appears effective in restoring linkage equilibrium in A. lyrata, as indicated by the rapid linkage disequilibrium decay around gene sequences (Wright et al., 2006; Kuittinen et al., 2007; Ross-Ibarra et al., 2008), polymorphisms may still remain locally structured over nearby nucleotide sites.
Accession codes
References
Al-Shehbaz IA, O’Kane SL (2002). Taxonomy and phylogeny of Arabidopsis (Brassicaceae). In: Somerville CR, Meyerowitz EM (eds) The Arabidopsis Book. American Society of Plant Biologist: Rockville, Maryland, pp 1–22.
Alsos IG, Engelskønt T, Gielly L, Taberlet P, Brochmann C (2005). Impact of ice ages on circumpolar molecular diversity: insights from an ecological key species. Mol Ecol 14: 2739–2753.
Ansell SW, Grundmann M, Russell SJ, Schneider H, Vogel JC (2008). Genetic discontinuity, breeding-system change and population history of Arabis alpina in the Italian Peninsula and adjacent Alps. Mol Ecol 17: 2245–2257.
Ansell SW, Schneider H, Pedersen N, Grundmann M, Russell SJ, Vogel JC (2007). Recombination diversifies chloroplast trnF pseudogenes in Arabidopsis lyrata. J Evol Biol 20: 2400–2411.
Beck JB, Schmuths H, Schaal BA (2008). Native range genetic variation in Arabidopsis thaliana is strongly geographically structured and reflects Pleistocene glacial dynamics. Mol Ecol 17: 902–915.
Casgrain P, Legendre P (2001). The R-package for Multivariant and Spatial Analysis. Version 4: 00 (d6) Départment de sciences biologiques, Université de Montréal Available at http://www.bio.umontreal.ca/casgrain/en/labo/R/v4/index.html.
Charlesworth D, Bartolome C, Schierup MH, Mable BK (2003). Haplotype structure of the stigmatic self-incompatibility gene in natural populations of Arabidopsis lyrata. Mol Biol Evol 20: 1741–1753.
Clauss MJ, Mitchell-Olds T (2006). Population genetic structure of Arabidopsis lyrata in Europe. Mol Ecol 15: 2753–2766.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Ehrich D, Gaudel M, Assefa A, Koch MA, Mummenhoff K, Nemomissa S, et al., Intrabiodiv Consortium (2007). Genetic consequences of pleistocene range shifts: contrast between the arctic, the alps and the east African mountains. Mol Ecol 16: 2545–2559.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S (2005). ARLEQUIN (version 3.0): An intregated software package for population genetic data analysis. Evol Bioinform Online 1: 47–50.
Gabrielsen TM, Bachmann K, Jakobsen KS, Brochmann C (1997). Glacial survival does not matter: RAPD phylogeography of Nordic Saxifraga oppositifolia. Mol Ecol 6: 831–842.
Gaudeul M, Stenønien HK, Ågren J (2007). Landscape structure, clonal proprogation, and genetic diversity in Scandinavian populations of Arabidopsis lyrata (Brassicaceae). Am J Botany 94: 1146–1155.
Hall TA (1999). BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program. North Carolina State University: Raleigh. Available at http://www.mbio.ncsu.edu/BioEdit/bioedit.html.
Hamrick JL, Godt MJW (1996). Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond, Ser B 351: 1291–1298.
Hejeny S, Slavik B (1992). Cardaminopsis petraea, Kvetena České Republiky. vol. 3. Academia Press: Praha, pp117–118.
Hemp A (1996). Okologie, Verbreitung und Gesellschaftsanschluβ ausgewhlter Eiszeitrelikte (Cardaminopsis petraea, Draba aizoides, Saxifraga decipiens, Arabis alpina und Asplenium viride) in der Pegnitzalb. Berichte der Bayrischen Botanischen Gesellschaft 66/67: 233–267.
Hewitt GM (2004). Genetic consequences of climate oscillations in the quaternary. Philos Trans R Soc Lond, Ser B 359: 183–195.
Ibrahim K, Nichols RA, Hewitt GM (1996). Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77: 282–291.
Jalas J, Suominen J (1994). Atlas florae Europaeae. Cruciferae (Symbrium to Aubrieta). vol. 10. Helsinki University Press: Helsinki, pp180–181.
Jonsell B, Kustas K, Nordal I (1995). Genetic variation in Arabis petraea, a disjunct species in northern Europe. Ecography 18: 321–332.
Kärkkäinen K, Kuittinen H, van Treuren R, Vogl C, Oikarien S, Savolainen O (1999). Genetic basis of inbreeding depression in Arabis petraea. Evolution 53: 1354–1365.
Kivimäki M, Kärkkäinen K, Gaudeul M, Loe G, Ågren J (2007). Gene, phenotype and function: Glabrous1 and resistance to herbivory in natural populations of Arabidopsis lyrata. Mol Ecol 16: 453–462.
Koch MA, Haubold B, Mitchell-Olds T (2000). Comparative evolutionary analysis of the chalcone synthase and alcohol dehydrogenase loci among different lineages Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol 17: 1483–1498.
Koch MA, Matschinger M (2007). Evolution and genetic differentiation among relatives of Arabidopsis thaliana. Proc Natl Acad Sci 104: 6272–6277.
Kuittinen H, Niittyvuopio A, Rinne P, Savolainen O (2007). Natural variation in Arabidopsis lyrata vernalization requirement conferred by a FRIGIDA indel polymorphism. Mol Biol Evol 25: 319–329.
Lang G (1994). Quartäre Vegetationsgeschichte Europas. Gustav Fischer: Jena.
Leinonen PH, Sandring S, Quilot B, Clauss MJ, Mitchell-Olds T, Rgen J et al. (2009). Local adaptation in Euorpean populations of Arabidopsis lyrata (Brassicaceae). Am J Botany 96: 1129–1137.
Lewis PO, Zaykin D (2001). Genetic Data Analysis: Computer program for the analysis of allelic data Version 1.1. Available at http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php.
Mable BK, Adam A (2007). Patterns of genetic diversity in outcrossing and selfing populations of Arabidopsis lyrata. Mol Ecol 16: 3565–3580.
Mitchell-Olds T (2006). Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 441: 947–952.
Mráz P, Gaudeul M, Rioux D, Gielly L, Choler P, Taberlet P, IntraBioDiv Consortium (2007). Genetic structure of Hypochaeris uniflora (Asteraceae) suggests vicariance in the Carpathians and rapid post-glacial colonization of the Alps from an eastern Alpine refugium. J Biogeogr 34: 2100–2114.
Muller M-H, Leppälä J, Savolainen O (2008). Genome-wide effects of postglacial colonization in Arabidopsis lyrata. Heredity 100: 47–58.
Nielsen R (2005). Molecular signatures of natural selection. Ann Rev Genet 39: 197–218.
Petit RJ, Aguinagalde I, de Baeulie JL, Bittkau C, Brewer S, Cheddadi R et al. (2003). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565.
Petit RJ, El Mousadik A, Pons O (1998). Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12: 844–855.
Piry S, Luikart G, Cornuet J-M (1999). BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Polataschek A (1966). Cytotaxonomische Beitrage zur Flora der Ostalpenländer, I. Österreichische Botanische Zeitschrift 113: 1–46.
Pritchard JK, Stephens M, Donelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Ratcliffe DA (1994). Arabis petraea In: Stewart A, Pearman DA, Preston CD (eds). Scarce plants of the British Isles. JNCC: Peterborough. pp 51.
Rogers AR, Harpending H (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9: 552–569.
Ross-Ibarra J, Wright SI, Foxe JP, Kawabe A, DeRose-Wilson L, Gos G et al. (2008). Patterns of Polymorphism and Demographic History in Natural Populations of Arabidopsis lyrata. PloS One 3: e2411.
Sandring S, Ågren J (2009). Pollinator-mediated selection on floral display and flowering time in the perennial herb Arabidopsis lyrata. Evolution 63: 1292–1300.
Schierup MH (1998). The effect of enzyme heterozygosity on growth in a strictly outcrossing species: the self-incompatible Arabis petraea (Brassicaceae). Hereditas 128: 21–31.
Schierup MH, Bechsgaard JS, Nielsen LH, Christiansen FB (2006). Selection at work in self-incompatible Arabidopsis lyrata: mating patterns in a natural population. Genetics 72: 477–484.
Schmickl R, Kiefer C, Dobeš C, Koch MA (2009). Evolution of trnF(GAA) pseudogenes in cruciferous plants. Plant Syst Evol 282: 229–240.
Stern R, Leidi S, Dale I, Grayer C (2005). INSTAT—an interactive statistical package. Statistical Services Centre, The University of Reading, UK. Available at http://www.rdg.ac.uk/ssc/software/instat/instat.html.
Taberlet P, Fumagalli L, Wust-Saucy A-M, Cosson J-F (1998). Comparative phylogeography and postglacial colonisation routes in Europe. Mol Ecol 7: 453–464.
van Treuren R, Kuittinen H, Karkkainen K, Baena-Gonzalez E, Savolainen O (1997). Evolution of microsatellites in Arabis petraea and Arabis lyrata, outcrossing relatives of Arabidopsis thaliana. Mol Biol Evol 14: 220–229.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wright SI, Lauga B, Charlesworth D (2003). Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol Ecol 12: 1247–1263.
Wright SI, Foxe JP, De-Rose-Wilson L, Kawabe A, Loosely M, Gaut BS et al. (2006). Testing for Effects of Recombination Rate on Nucleotide Diversity in Natural Populations of Arabidopsis lyrata. Genetics 174: 1421–1430.
Yeh FC, Yang R, Boyle T (1999). POPGENE Microsoft Window-based Freeware for Population Genetic Analysis Version 1 31 Centre for International Forestry Research, University of Alberta. Available at http://www.ualberta.ca/∼fyeh/.
Acknowledgements
We thank a number of friends and collaborators who contributed to field sampling: the late John Barrett, Juliet Brodie, Paul Harvey, Joseph Greimler, Starri Heimarsson, Svante Holm, Stefan Jessen, William Kunin, Helena Persson, Daniel Stancik and staff of the UK national conservation agencies SNH and CCW, especially Chris Sydes and Barbara Jones, and also Anne-Marie Van Dodeweerd who kindly proof-read this article. This work was financially supported by a NERC PhD studentship to SWA and the NERC Grant GR3/12073. SWA also received a travel grant from the BSBI.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Ansell, S., Stenøien, H., Grundmann, M. et al. Population structure and historical biogeography of European Arabidopsis lyrata. Heredity 105, 543–553 (2010). https://doi.org/10.1038/hdy.2010.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.10
Keywords
This article is cited by
-
Interspecific introgression mediates adaptation to whole genome duplication
Nature Communications (2019)
-
An Arabidopsis introgression zone studied at high spatio-temporal resolution: interglacial and multiple genetic contact exemplified using whole nuclear and plastid genomes
BMC Genomics (2017)
-
R-gene variation across Arabidopsis lyrata subspecies: effects of population structure, selection and mating system
BMC Evolutionary Biology (2016)
-
The structure of allozyme variation in Silene nutans (Caryophyllaceae) in Denmark and in north-western Europe
Plant Systematics and Evolution (2016)
