
Abstract
An increasing number of phylogeographic studies on marine species shows discordant patterns in the degree of population differentiation between nuclear and mitochondrial markers. To understand better which factors have the potential to cause these patterns of discordance in marine organisms, a population genetic study was realized on the sand goby Pomatoschistus minutus (Pallas 1770; Gobiidae, Teleostei). Sand gobies from eight European locations were genotyped at eight microsatellite markers. Microsatellites confirmed the global phylogeographical pattern of P. minutus observed with mitochondrial DNA (mtDNA) markers and nuclear allozyme markers. Three groups consistent with the mitochondrial lineages were defined (the Mediterranean, Iberian and North Atlantic groups) and indications of a recent founder event in the northern Baltic Sea were found. Nevertheless, differences in the degree of population differentiation between the nuclear and mitochondrial markers were large (global FST-values for microsatellites=0.0121; for allozymes=0.00831; for mtDNA=0.4293). Selection, sex-biased dispersal, homoplasy and a high effective population size are generally accepted as explanations for this mitonuclear discrepancy in the degree of population differentiation. In this study, selection on mtDNA and microsatellites, male-biased dispersal and homoplasy on microsatellite markers are unlikely to be a main cause for this discrepancy. The most likely reason for the discordant pattern is a recent demographical expansion of the sand goby, resulting in high effective population sizes slowing down the differentiation of nuclear DNA.
Similar content being viewed by others

Introduction
Traditionally, the genetic structure of marine organisms has been thought to be homogeneous because of the lack of obvious barriers to gene flow in the ‘open’ marine environment. In the last decade, however, an increasing number of population genetic studies have described distinct genetic structuring for several marine species on large and small geographical scales (Hauser and Carvalho, 2008). Those observed population genetic structures reflect both historical and contemporary processes (Balloux and Lugon-Moulin, 2002). Geographical and climatic factors acting during the Pleistocene glaciations (1800–11.5 ka) are the major factors responsible for the present genetic structure of most extant marine species (Hewitt, 2000). Heterogeneity in the marine environment owing to the influence of climate, hydrodynamics and topography, together with biological traits, such as sex-dependent migration, phylopatry and assortative mating, which may counteract gene flow, enhance genetic structuring (Ruzzante et al., 1998).
These recent insights are mainly due to the increased popularity of polymorphic microsatellite markers in marine population and landscape genetics (Jørgensen et al., 2005). Microsatellites have proven for many species to be more powerful for resolving population structure than mitochondrial DNA (mtDNA) and allozyme markers (De Innocentiis et al., 2004; Nesbø et al., 2000). However, this is not applicable to all marine organisms (Lukoschek et al., 2008). There is almost certainly a publication bias for marine studies detecting microscale population structures with microsatellites (Hauser and Carvalho, 2008). An increasing number of studies have shown a large difference in the order of magnitude for the population divergence between nuclear and mitochondrial markers. Among those, mtDNA shows a higher sensitivity to resolve the phylogeographical and population genetic structure (Peijnenburg et al., 2006; Lukoschek et al., 2008). This discordance in resolution among markers may result from the differential effects of genetic drift, mutation and migration on a marker class, or may result from selection or sex-biased dispersal (Buonaccorsi et al., 2001). More research is required to study how common and important those factors are in the marine environment.
The sand goby Pomatoschistus minutus (Pallas 1770; Gobiidae, Teleostei) is a small marine demersal fish common in shallow waters along European coasts (Miller, 1986) (Figure 1). Although earlier analyses with allozyme markers showed very low population differentiation values for P. minutus (Stefanni et al., 2003), a recent study revealed that the sand goby is highly structured at the mtDNA cytochrome b (cyt b) locus (Larmuseau et al., 2009b). On the basis of mtDNA, middle Pleistocene glaciations yielded three isolated and different evolving sets of sand goby populations. Reciprocal monophyly was observed between a Mediterranean Sea (MS) and an Atlantic Ocean (AO) Clade. The AO-Clade contains two evolutionary significant units: the Iberian Peninsula (IB) Group and the North Atlantic Group. For the North Atlantic Group, there is evidence for geographic sorting of the ancestral mtDNA haplotypes with recent independent radiations in the Baltic Sea, Irish Sea, North Sea and the Bay of Biscay (Larmuseau et al., 2009b). Analyses with allozyme markers revealed weak divergence between Mediterranean and Atlantic populations for P. minutus and no differentiation elsewhere, which was attributed to high contemporary gene flow throughout its whole distributional range (Stefanni et al., 2003). However, the biology and morphology of sand gobies indicate reduced levels of contemporary gene flow (Miller, 1986). Moreover, the mitochondrial phylogeography invalidates the interpretation of the allozyme data because of the high FST values between sand goby populations and the lack of common haplotypes between the three evolutionary significant units (Larmuseau et al., 2009b). The reasons for this discordant pattern can be surveyed by using another set of nuclear markers, such as DNA microsatellites. In contrast to allozymes, DNA microsatellite markers promise a higher resolution for population differentiation in the sand goby because of a higher mutation rate (Chistiakov et al., 2006). Moreover, Pampoulie et al. (2004) detected genetic structure on a microscale for P. minutus in the southern North Sea using microsatellites, whereas Gysels et al. (2004b) did not in the same region using allozyme markers.

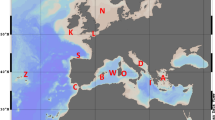
Geographical distribution of the eight sampling locations of sand goby P. minutus. The shaded area represents the distribution range of P. minutus according to Miller (1986). The two dashed lines represent the main phylogeographical barriers for the sand goby based on the mtDNA analysis of Larmuseau et al. (2009b) that differentiate the three isolated and differently evolving sets of populations. See Table 1 for sample codes.
In this study, two hypotheses are tested: (a) is the phylogeographical structure of the sand goby based on data of the mtDNA cyt b locus (Larmuseau et al., 2009b) confirmed by nuclear microsatellite markers, and (b) do microsatellite markers in comparison with mitochondrial markers show a superior resolution to reveal the phylogeographical and population genetic structure?
Materials and methods
Sampling and species/sex identification
A total of 696 P. minutus individuals were caught at eight locations along the European coast between September 2002 and January 2007 (Table 1 and Figure 1). Many of the samples have already been included in a previous mtDNA study (Larmuseau et al., 2009b). Locations BNS and VMS were sampled twice to perform an estimation of effective population size. All new samples were identified as P. minutus morphologically, based on the dermal head papillae (Miller, 1986) and pigmentation pattern (Hamerlynck, 1990), and genetically according to a molecular tool described by Larmuseau et al. (2008). The sex of each sand goby was determined by the shape of the urogenital papilla as drawn by Rodrigues et al. (2006).
Microsatellite genotyping
Genomic DNA was extracted from fin clips stored in 100% ethanol using the NucleoSplin Extraction Kit (Machery-Nagel GmBH, Düren, Germany). Each individual was genotyped at nine multiplexed microsatellite loci (Pmin03, Pmin04, Pmin09, Pmin16-2, Pmin20, Pmin29, Pmin31, Pmin35 and Pmin38) (Larmuseau et al., 2007) on an ABI 3130 automated capillary DNA sequencer (Applied Biosystems, Foster City, CA, USA). Several methods were used to mitigate genotyping errors in the data set (Supplementary materials); marker Pmin20 was excluded for further statistical analyses.
Genetic diversity
We used genotype and allele frequencies of the microsatellite loci to obtain standard estimates of genetic diversity within and between sample sites. Genetic variation in each population was measured by calculating the mean number of alleles per locus, the observed (HO) and unbiased expected (HE) heterozygosities and the FIS. The deviation from the Hardy–Weinberg equilibrium was assessed with GENETIX v.4.05 (Belkhir et al., 2004). Allelic richness, which corrects the number of alleles for sample size, was assessed using FSTAT v.2.9.3.2 (Goudet, 2001). Exact tests of linkage disequilibrium between pairs of loci were calculated at each location, each region and across all samples using GENEPOP v.3.4. (Raymond and Rousset, 1995).
Patterns of population subdivision
Different methods were used to reveal the population substructure of P. minutus. First, a factorial correspondence analysis (FCA) of individual multilocus genotypes was performed using GENETIX to reveal the portion of the hyperspace of all genotypes occupied by each group of individuals. Second, population differentiation was quantified in GENETIX using the standardized allelic variation FST, estimated as θ (Weir and Cockerham, 1984), and in SPAGeDi 1.2 g (Hardy and Vekemans, 2002) using an analog of FST for microsatellites RST, estimated as ñ (Slatkin, 1995). FST-linked pairwise genetic distances were calculated according to Cavalli-Sforza and Edwards (1967) (DCE) with GENETIX, and RST-linked pairwise genetic distances calculated according to Goldstein et al. (1995) (dμ2) were obtained with SPAGeDi. FST value and DCE value were tested for significance against 104 random permutations of the data in GENETIX. The significance for RST value and dμ2 value were tested in SPAGeDi also against 104 random permutations. A sequential Bonferroni test (Rice, 1989) and the false discovery rate control (Verhoeven et al., 2005) were applied to correct significance levels for multiple testing. Standardized genetic differentiation measures were obtained by dividing FST measures by the maximum values for FST (Hedrick, 2005; Meirmans, 2006), calculated using the pragmatic recoding approach suggested by Meirmans (2006). To assess the influence of stepwise-like mutations versus drift on genetic differentiation, we performed a permutation test available in the software SPAGeDi. Allele size at each locus was randomly permutated among allelic states (2000 mutations) to simulate a distribution of RST values (pRST) and 95% confidence intervals (CIs) under the null hypothesis that differences in allele sizes do not contribute to population differentiation (Hardy et al., 2003). Third, the classical multidimensional scaling analyses based on the two types of genetic distances were obtained using the Vegan package in R (Oksanen et al., 2007). Ordination plots with a stress value below 0.20 provide interpretable information concerning intersite relationships (Clarke, 1993). Fourth, to analyze the effect of geographical distance on genetic distance, the Mantel test in GENETIX (Belkhir et al., 2004), which computes the correlation between distance matrices by means of a permutation procedure (Mantel, 1967; Smouse et al., 1986), was used. Geographical distances were obtained as the shortest coastal distances between sites using the electronic atlas ENCARTA (Microsoft 2001). Both types of pairwise genetic distances, DCE and dμ2, were used for the Mantel test. Fifth, a Bayesian clustering analysis was realized for the microsatellite data using the program STRUCTURE v.2.2 (Pritchard et al., 2000). This approach, which estimates the number of independent genetic clusters in the data set, does not require a priori information about population structure, and thus provides an estimate of genetic structure independent of the origin of samples. We used the no-admixture algorithm without prior population information and used 10 000 runs as burn-in and 100 000 runs for each of three Markov chains. The no-admixture algorithm was used on the basis of detection of the lack in gene flow between and within marine systems for P. minutus, at the mtDNA level (Larmuseau et al., 2009b). Moreover, this model is appropriate for studying fully discrete populations and is often more powerful than the admixture model at detecting subtle structure (see manual STRUCTURE v.2.2). For each simulation of k=1–10 (no. of clusters), we used 10 replicates. The uncorrelated allele frequency model with parameter λ set to 1 was used as the model assuming that the allele frequencies in each population are independent. This is the original model used in Pritchard et al. (2000). We selected the most likely number of clusters given the data by choosing the number of clusters where we observed the largest difference in log likelihoods (ΔK) (Evanno et al., 2005). Finally, the overall pattern of population genetic structure among regions was assessed with a Bayesian approach developed by Ciofi et al. (1999) and implemented in the program 2MOD by MA Beaumont (http://www.rubic.rdg.ac.uk/~mab/software.html). Two models are evaluated: (1) the gene flow model, which assumes that gene frequencies within populations are caused by a balance between genetic drift and gene flow, and (2) the drift model, in which allele frequencies within populations are evolving purely through drift. The Markov chain Monte Carlo search was carried out twice using 105 iterations. The first 10% of the runs were discarded to remove the effects of initial starting parameters. In each case, the probability of a model was estimated from the number of times it appeared during the simulation.
Comparison between microsatellite and mitochondrial data
MtDNA cyt b data were available for eight out of ten sand goby samples (TBS, WCS, RNS, BNS1, WIS, GOA, AAO and VMS1) (Larmuseau et al., 2009b). On the other hand, the allozyme analyses by Gysels (2003) and by Stefanni et al. (2003) were restricted to samples from other locations. Therefore, it was only possible to compare statistically the results of microsatellites and mtDNA cyt b data. Two statistical methods were used to compare the degree of population differentiation between the types of genetic distances calculated for the microsatellite data (DCE and dμ2) and the genetic distances of Tamura and Nei (1993) calculated for the mtDNA data. Data on the mtDNA cyt b were available for the populations. First, the pairwise FST matrices were correlated by using simple Mantel procedures (Mantel, 1967) in the Vegan library in R (Oksanen et al., 2007). Permutations (n=10 000) were used to evaluate statistical significance. Then, two-dimensional multidimensional scaling analysis ordinations of the two types of genetic marker were compared by Procrustes analysis (Gower, 1975) using R software. Procrustes analysis is searching for the best match between two configurations of points in a multivariate Euclidean space using rotation, translation, reflection and dilation of one configuration. The criterion used to assess the best fit is the minimization of the sum of squares between the differences for each observation (m2). The significance of the result, an optimal superposition of one configuration on the other (reference), is obtained through a permutation test (PROTEST) (Jackson, 1995). PROTEST is using R=sqrt(1−m2) as a test statistic, which can be interpreted as a correlation.
Impact of selection, sex-biased dispersal and effective population size on the degree of population divergence estimates
To verify if the variation on the microsatellites can be influenced by selection, two different approaches were considered. First, all microsatellite flanking regions were compared with sequences in GenBank by means of the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/) (Altschul et al., 1990) to verify if the microsatellites potentially are located within the described functional regions of the genome. Second, potential outlier microsatellites were identified by using the selection detection workbenches LOSITAN (Antao et al., 2008) and BayeScan (Foll and Gaggiotti, 2008). Analyses are performed for LOSITAN with 10 000 simulations for IAM and SMM, both with the options ‘Neutral’ mean FST and force mean FST. For BayeScan, 10 000 iterations were conducted with a thinning interval of 20 and with 10 pilot runs.
To detect differences in migration rates between females and males, deviation from the Hardy–Weinberg equilibrium (FIS), differentiation among populations (FST), relatedness (r) (Queller and Goodnight, 1989), mean assignment index (mAIC) and variance of the assignment index (vAIC) were quantified separately for both sexes over all populations (Goudet et al., 2002). Statistical significance of differences in these within-population indices was determined with 10 000 permutations using the randomization method implemented in FSTAT (Goudet, 2001).
Effective population sizes were estimated using different point methods, which do not require samples spaced over at least one temporal interval, and temporal methods, where samples are taken from the same location at two or more points in time separated by a specified number of generations (one or more). Two different point estimation methods were estimated for the 10 samples. The program NeEstimator v.1.3 (Peel et al., 2004) was used for the Heterozygote Excess method, which examines the excess of heterozygotes in the sample compared with the proportion predicted under the Hardy–Weinberg equilibrium (Luikart and Cornuet, 1999). For the estimation of effective population size on the basis of linkage disequilibrium data, the program LDNe v.1.31 (Waples and Do, 2008) was used because it implements a recently developed bias correction (Waples, 2006). Finally, Ne was estimated using two temporal methods in NeEstimator v.1.3: (i) based on the Moment Approach (Waples, 1989) and (ii) on the pseudo-likelihood method (MLNE) developed by Wang and Whitlock (2003). These two methods require at least two temporally spaced samples, and therefore this analysis was only possible for VMS and BNS (Table 1).
Results
Genetic diversity
Mean allelic richness per location, corrected for sample size, varied between 12.088 (northern Baltic Sea) and 15.454 (MS) (mean=13.622) (Table 2). Mean expected heterozygosity was relatively uniform among the different sampling sites with the lowest value in the northern Baltic Sea sample (0.740) and the highest in the MS samples (0.781) (mean=0.756) (Table 2). Locations WCS and BNS2 showed a significant departure from the Hardy–Weinberg equilibrium (Table 2) (both multilocus FIS=0.050). Pairwise comparisons between loci revealed no significant linkage disequilibrium after sequential Bonferroni corrections.
Patterns of population subdivision
First, the graphical distribution of populations from the FCA showed that the two Mediterranean samples (VMS1 and VMS2), as well as all Atlantic samples, except the one from the IB (AAO), clustered together (Figure 2). Disregarding the AAO sample, the sample of the Bay of Biscay (GOA) was the most aberrant among the Atlantic samples. The distribution of all individuals in a FCA plot (graph not shown) shows a small overlap between the Mediterranean and Atlantic samples as well as between AAO and the other Atlantic samples.

Factorial correspondence analysis plot based on microsatellite data of the 10 samples of P. minutus. See Table 1 for sample codes.
Second, the global θ- and ρ-values across all samples, excluding the temporal samples (BNS2 and VMS2), were 0.0121 and 0.0426, respectively (both highly significant, P<0.001). The global standardized multilocus FST was 0.0484. The pairwise FST values were significant after sequential Bonferroni correction and false discovery rate control for all population pairs including samples TBS, VMS and AAO, together with a limited number of population pairs between Atlantic populations (Table 3). Pairwise RST values were significant after sequential Bonferroni correction or false discovery rate control for several population pairs between VMS and Atlantic populations (Table 3). The pairwise FST and RST values between the two temporal samples were not statistically different (P>0.05) for BNS (0.0038 and 0.0005, respectively) and VMS (−0.0012 and −0.0025, respectively) (Table 3). The pairwise standardized FST values between the different samples are listed in Table 4. Jackknife analysis revealed that locus Pmin16-2 was responsible for the largest divergence as calculated with RST, but not with FST (average pairwise RST=0.02974 with locus Pmin16-2 and RST=0.00528 without this locus). Random permutation of different allele sizes among allelic states at each locus revealed that estimates of RST were significantly larger than the 95% CI range of the pRST values at one single locus Pmin16-2 (Table 5), suggesting a mutational component to genetic differentiation. For the other microsatellites, neither loci allele size nor stepwise mutations strongly influence the population differentiation of sand gobies. Hardy et al. (2003) suggest that, in this situation, FST should be preferred over RST for estimating population differentiation.
Third, the classical multidimensional scaling analyses plots based on both types of genetic distances separated clearly the group of the two Mediterranean samples from the populations of the Atlantic (Figure 3). Both plots had a stress value below 0.20, suggesting interpretable information concerning intersite relationships.

Classical multidimensional scaling plots of pairwise genetic distance for the microsatellite data calculated according to (a) Cavalli-Sforza and Edwards (1967) (DCE) and (b) Goldstein et al. (1995) (dμ2) among the 10 samples of P. minutus. See Table 1 for sample codes.
Fourth, the global Mantel test revealed a significant isolation by distance pattern with DCE (r=0.713, P<0.05) but not with dμ2 (r=0.166, P>0.05). However, no significant isolation by distance was found for both types of genetic distances when the MS and/or IB samples were excluded from the data set.
Fifth, testing the significance of the stepwise clustering procedure performed in STRUCTURE resulted in separation of the samples into three hypothetical clusters (highest ΔK for K=3). The lowest proportion for a sample of membership to a particular cluster is 0.411 in cluster 1 for sample WCS and the highest proportion is 0.897 in cluster 2 for sample TBS (Table 6). All Atlantic samples (excluding TBS) had the highest assignment value for cluster 1; the northern Baltic sample (TBS) for cluster 2 and the two Mediterranean samples for cluster 3 (Table 6 and Figure 4).

Results of the clustering analysis conducted in STRUCTURE 2.2 (Pritchard et al., 2000) based on the microsatellite data. In the bar plot, each of the 696 individuals is represented by a vertical bar indicating its estimated proportion of membership to the three clusters (K=3). See Table 1 for sample codes.
Finally, the Bayesian approach used in 2MOD indicated that P. minutus populations were at migration-drift equilibrium. All sampled iterations revealed that a migration-drift equilibrium model was more likely than the non-equilibrium drift model.
Comparison between types of genetic distances and mtDNA
FST-values are much lower for microsatellites (global FST=0.0121 (CI=0.0066–0.0174) and global standardized FST=0.0484 (CI=0.0264–0.0696), both without the temporal samples VMS2 and BNS2) than for mtDNA (global FST=0.4293 (CI=0.3231–0.5302)). The proportion of differences in FST between mtDNA and nuclear microsatellites is not attributed to stochastic variability among loci as there were no notable differences between different microsatellite loci. The highest global θ was found for the locus Pmin 16-2 (0.0314); the lowest θ-value was found for the locus Pmin 38 (−0.0023). Pairwise FST values between all samples based on the mtDNA data are presented together with the standardized pairwise FST estimates for microsatellites in Table 4. Each pairwise FST value for mtDNA is on average 10 times higher than the value for microsatellites. The pairwise genetic distances of the microsatellite data (dμ2 and DCE) correlated well with each other (Procrustes R=0.8339, P-value=0.004; Mantel R=0.8574, P-value <0.001). Procrustes analyses and Mantel tests also showed that single-locus dμ2 and DCE correlated well with the estimates based on all microsatellites. The DCE pairwise genetic distances of the microsatellite data correlated well with Tamura and Nei (1993) genetic distances based on the mtDNA data (Procrustes R=0.746, P-value <0.001; Mantel R=0.6804, P-value=0.032). In contrast, the dμ2 pairwise genetic distances of the microsatellite data did not correlate significantly with the distances based on the mtDNA data (Procrustes R=0.660, P-value=0.086; Mantel R=0.645, P-value=0.081).
Impact of selection, sex-biased dispersal and effective population size on the degree of population divergence estimates
No indication for selection for some of the microsatellite markers was observed in the analysis. The results of Lositan and BayeScan were congruent with this pattern by revealing no outlier loci. Moreover, a blast search of the flanking regions of the microsatellite loci in de GenBank database revealed that they are not linked to a functional region already sequenced for any organism available in GenBank.
A total of 352 males and 344 females were genotyped and the population genetic parameters were estimated separately for each sex. No parameter was significantly different between the sexes (Table 7), suggesting no sex-biased dispersal.
Estimates from the various point and temporal methods to estimate the Ne are not congruent with each other (Table 8). Both point methods cannot exclude the possibility that the population sizes are infinite. The two temporal methods are similar for the BNS population (∼400 individuals) and for VMS (>1000 individuals).
Discussion
Phylogeography of the sand goby
The current study with microsatellites revealed a subtle but significant genetic structure within P. minutus. The highest divergence in the analysis was found between populations of the Atlantic, comprising all sand goby populations spanning from the IB to the Baltic Sea, and the MS, represented by individuals from the Vaccarès lagoon (Gulf of Lion) (Table 3 and Figures 2, 3 and 4). The microsatellite marker Pmin16-2 shows a clear differentiation with a small overlap in allele size between the MS-Clade and AO-Clade, clearly indicating the historical divergence of both regions. In addition, the highest divergence noticed in the genetic analysis of allozyme markers were also found between Mediterranean and Atlantic sand gobies (Stefanni et al., 2003; Larmuseau et al., 2009b). The results are also congruent with the phylogenetic analysis based on mtDNA cyt b, which clearly showed two monophyletic clades within P. minutus, the Mediterranean Sea Clade (MS-Clade) and the AO-Clade (Larmuseau et al., 2009b). Divergence between the two sand goby clades is most likely the result of geographic isolation caused by one of the Middle Pleistocene sea-level drops (Larmuseau et al., 2009b). The Mediterranean populations showed the highest variability in the microsatellite markers (Table 2) despite the small distribution range along the northwestern MS coasts and lagoons (Miller, 1986) (Figure 1). Larmuseau et al. (2009b) noticed the highest diversity in mtDNA in the Mediterranean samples as well. Furthermore, the highest species diversity of the ‘sand goby’ group is observed within the MS (Huyse et al., 2004) supporting a Mediterranean origin of P. minutus, as suggested by Gysels et al. (2004a). Huyse et al. (2004) estimated the origin of the species between 1.94 and 1.18 mya (early Pleistocene). The Pleistocene glaciations were the most significant historical events during the evolutionary lifespan of most Holarctic species and are believed to have accelerated the speciation process in the present day sister taxa (Avise, 2000).
Limited genetic differentiation was observed between the Atlantic samples with microsatellites, but the FCA and the pairwise FST values suggest that the IB's sample is different from those of all other Atlantic populations (Figure 2 and Table 3). These results are congruent with the two mitochondrial groups within the Atlantic samples: the Iberian (IB-Group) and the North Atlantic Group. This divergence is likely the result of the population decline within different refugia during glaciations in the Middle Pleistocene (Larmuseau et al., 2009b). On the basis of the Bayesian assignment test and pairwise FST values, the position for the sample of the northern Baltic Sea (TBS) was remarkable (Tables 3, 6 and Figure 4). This is most likely the result of a founder event, based on the low variation on the microsatellite loci in this population (Table 2) and as suggested by Larmuseau et al. (2009b) based on the mtDNA data. The difference in the cyt b haplotype network between the southern and northern Baltic Sea samples suggested that the Baltic Sea has been colonized in two phases over a period of 8000 years, with a stronger founder effect in the north. The presence of only two common cyt b haplotypes in the northern samples HBS and TBS (haplotypes NA01 and NA28) with their many uniquely derived haplotypes suggested that only a few individuals founded the northern Baltic Sea (Johannesson and André, 2006). A recent study has already shown that sand gobies are locally adapted to the specific light environment of the northern Baltic Sea in comparison with the sand gobies of the North Sea and the AO (Larmuseau et al., 2009a). If only a few sand gobies colonized the northern Baltic, the populations in this region will be genetically diverged from the founding population, mainly through a shift in allele frequencies. Such shifts are detectable with pairwise FST values and the STRUCTURE analysis (Table 3 and Figure 4). In contrast, individual multilocus genotypes are less sensitive for shifts in allele frequencies, and analyses based on multilocus genotypes, such as the FCA plot (Figure 2), will not be able to distinguish between different groups after a founding event.
The Mantel test and Procrustes analysis illustrated that the microsatellite results are congruent with the mtDNA phylogeographic pattern. However, those tests were not significant when the dμ2 genetic distance was used for microsatellites. This suggests that, although both measures are correlated, FST correlates better with mtDNA than RST. In theory, FST is more sensitive than RST for recent intraspecific divergence (Gaggiotti et al., 1999; Balloux and Lugon-Moulin, 2002). Moreover, RST can be less accurate in reflecting population differentiation because of its higher associated variances (Balloux and Lugon-Moulin, 2002). Therefore, the number of loci screened has to increase before a consistent pattern is reached (Gaggiotti et al., 1999; Balloux and Goudet, 2002). This has already been empirically observed with European grayling Thymallus thymallus (Koskinen et al., 2004).
Overall, the results confirm the first hypothesis of this study that stated that microsatellite markers reveal a phylogeographic pattern congruent with the patterns based on mtDNA and allozyme data.
Differences in the degree of divergence estimates among nuclear and mtDNA markers
The difference in the degree of population differentiation between the nuclear and mitochondrial markers is remarkable. The mitochondrial differentiation values are more than an order of magnitude higher than the nuclear differentiation, even after standardization of the microsatellite results. The standardized measure allows comparison between loci with different levels of genetic variation (Hedrick, 2005). Therefore, several other factors can be the cause of the observed discrepancy.
First, selection may have differential effects on genetic markers. It has been suggested that balancing selection may significantly influence the distribution of allozyme diversity (De Innocentiis et al., 2001). Loci experiencing balancing selection will have allele frequencies more similar than expected under neutrality, reducing the FST estimates. Allendorf and Seeb (2000) concluded that estimates of population structure produced by allozymes were generally comparable to those obtained with other nuclear markers, including microsatellites. They noted that when differences between marker classes did occur, they were usually due to one or a few exceptional loci and not all of them. Each used microsatellite marker was tested for positive or balanced selection in comparison with the other markers in the Lositan and BayeScan analysis, but no marker seemed to be an outlier. Similar to microsatellites, it is also not safe to assume a priori that mtDNA evolves as a strictly neutral marker (Ballard and Whitlock, 2004). Selection on mtDNA may accelerate the coalescence of lineages, and thus increase the levels of differentiation observed between populations (Peijnenburg et al., 2006). The observation of very low nuclear differentiation in sand gobies while mtDNA data revealed no gene flow between the three isolated Middle Pleistocene lineages cannot be explained by selective evolution for mtDNA. The rejection of the null hypothesis in different neutrality tests for the mtDNA data of P. minutus was assigned to demographical expansions instead of selection (Larmuseau et al., 2009b). Various demographic analyses on the mtDNA data showed that the intra-assemblage genetic structure of P. minutus contains signatures of demographic expansion events.
Second, studies documenting a weaker population subdivision for nuclear than maternally inherited genetic markers often attribute these discrepancies to male-biased dispersal. Sex-biased dispersal is common in nature (Cano et al., 2008); however, it has only been described for a limited number of marine fishes (Consuegra and de Leaniz, 2007). No indications for sex-biased dispersal were found for P. minutus with microsatellite markers (Table 7). Contemporary gene flow between the Mediterranean and Atlantic basins is also unlikely because of the discontinuous distribution of the sand goby (Miller, 1986) (Figure 1). Moreover, only males have to migrate successfully to explain the pattern. However, females are expected to be the most mobile sex, especially during the spawning period when males are guarding their nest (Lindström et al., 2006).
Third, various technical problems, including homoplasy, may have reduced the signal of differentiation detected by the microsatellite markers. Homoplasy occurs when different copies of a locus are identical in state, although not identical by descent. The situations where size homoplasy is most prevalent involve high mutation rates and large population sizes together with strong allele size constraints (Estoup et al., 2002). Therefore, effects of homoplasy are expected to be common for microsatellites in marine fishes (O’Reilly et al., 2004), which has implications for the identification of genetic structuring (Carreras-Carbonell et al., 2006). Microsatellites probably suffered from higher levels of homoplasy than mtDNA because of higher mutation rates and larger effective population sizes (Balloux et al., 2000). The various microsatellite markers of P. minutus indicate homoplasy because of the high allele numbers and the limited size range of all markers (on average 1 allele per 2.053 bp). Single-nucleotide polymorphisms and microsatellite markers with less alleles covering a limited size range might be useful to understand the power of homoplasy in the analysis. However, simulation studies suggest that size homoplasy will have much less effect on estimates of population differentiation than gene migration or genetic drift (Estoup et al., 2002). Therefore, it is unlikely that homoplasy is the main cause of the observed differences.
Finally, mitochondrial markers can be more sensitive in detecting differentiation because of a lower effective population size than nuclear markers (Shaw et al., 2004). Genetic drift effects are linked to effective population size (Ne), and therefore it is possible that an ecologically relevant population structure remains undetectable by using neutral markers when the Ne is high (Bentzen, 1998). Marine fish have the potential to have high Ne, and therefore recently separated large populations may appear genetically homogeneous even in the complete absence of contemporary gene flow (Hauser and Carvalho, 2008). Simulations in the study by Buonaccorsi et al. (2001) showed that differences in the magnitude of estimated population subdivision from nuclear and mitochondrial markers could be accounted for entirely by differences in effective population sizes and polymorphisms on FST estimates. The haploid and maternal-only inheritance of mtDNA has an effective population size of one-quarter that of nuclear DNA, making it more susceptible to effects of genetic drift (Shaw et al., 2004). This explanation has been invoked for the discordant patterns in population differentiation between nuclear and mitochondrial markers for marine organisms, such as the blue marlin Makaira nigricans (Buonaccorsi et al., 2001), Patagonian toothfish Dissostichus eleginoides (Appleyard et al., 2002; Shaw et al., 2004) and the olive sea snake Aipysurus laevis (Lukoschek et al., 2008). Simulations in EASYPOP v.1.7 (Balloux, 2001) with specific biological information about the sand goby showed no difference with the simulations of the study by Buonaccorsi et al. (2001) (results not shown). Moreover, P. minutus is known to be one of the most abundant fish species across almost its full range (Pasquaud et al., 2004; Ehrenberg et al., 2005; Maes et al., 2005). Therefore, high Ne most likely explains the discordant patterns between nuclear and mitochondrial data for the sand goby. Point methods to estimate the Ne of the present sampled populations cannot invalidate the null hypothesis of an infinite population size for P. minutus (Table 8). Nevertheless, point methods are not always reliable and are biased (Wang and Whitlock, 2003), especially when the sample size is small (<100 individuals) and below the true Ne (England et al., 2006). Both temporal Ne estimates show a limited population size, especially for the BNS location (100–1000 individuals) (Table 8). Still, there are crucial differences between the two temporal methods and the assumption of a closed system without migration could not be fulfilled. Migration inside the marine system can therefore cause a strong underestimation of the Ne (Wang and Whitlock, 2003). On the other hand, the high number of alleles for microsatellites confirms the hypothesis of a high effective population size of more than one thousand individuals to maintain the high genetic variation (Ewens, 1972; Poulsen et al., 2006). The Pmin20 locus was excluded from the analysis because, after genotyping more than 90 individuals, the number of alleles almost matched the number of fish genotyped. The studies of Jones et al. (2001) and Pampoulie et al. (2004) also observed microsatellite markers with a very high number of alleles. By organizing a better sampling strategy and using straightforward Ne-estimation methods incorporating migration (Wang and Whitlock, 2003), better estimates of the Ne might confirm the expected high Ne for P. minutus.
Our present results do not confirm the second hypothesis, stating that microsatellite markers are more sensitive for population differentiation on a macroscale than mitochondrial and allozyme markers. The most likely reason for the discordant pattern between nuclear and mitochondrial loci is that the recent demographical expansion in P. minutus resulted in high effective population sizes, slowing down the differentiation at nuclear loci.
Conclusion
Marine organisms have a high potential for gene flow and population size. However, it is not known whether the observed low genetic differentiation for so many marine species reflects high effective population sizes and low gene flow, high effective population sizes and high rates of gene flow, or low effective population sizes and high rates of gene flow (Hauser and Carvalho, 2008). In this study on P. minutus, the scenario of high effective population sizes and low gene flow best explains the observed genetic pattern. However, this conclusion could only be reached with more than one genetic marker. Therefore, one has to be cautious when interpreting the present-day genetic structure in terms of gene flow while using one type of marker or statistical method.
References
Allendorf FW, Seeb LW (2000). Concordance of genetic divergence among sockeye salmon populations at allozyme, nuclear DNA, and mitochondrial DNA markers. Evolution 54: 640–651.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008). LOSITAN: a workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinformatics 9: 323.
Appleyard SA, Ward RD, Williams R (2002). Population structure of the Patagonian toothfish around Heard, McDonald and Macquarie Islands. Antarct Sci 14: 364–373.
Avise JC (2000). Phylogeography: the History and Formation of Species. Harvard University Press: Cambridge, MA.
Ballard JWO, Whitlock MC (2004). The incomplete natural history of mitochondria. Mol Ecol 13: 729–744.
Balloux F (2001). EASYPOP (version 1.7): a computer program for population genetics simulations. J Hered 92: 301–302.
Balloux F, Brunner H, Lugon-Moulin N, Hausser J, Goudet J (2000). Microsatellites can be misleading: an empirical and simulation study. Evolution 54: 1414–1422.
Balloux F, Goudet J (2002). Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Mol Ecol 11: 771–783.
Balloux F, Lugon-Moulin N (2002). The estimation of population differentiation with microsatellite markers. Mol Ecol 11: 155–165.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004). GENETIX 4.05, Logiciel Sous WindowsTM Pour la Génétique Des Populations. Université de Montpellier II: Montpellier (France).
Bentzen P (1998). Seeking evidence of local stock structure using molecular genetic methods. In: Hunt von Herbing I, Kornfield I, Tupper M and Wilson J (eds). The implications of localized fisheries stocks. Regional Agricultural Engineering Service: New York, pp 20–30.
Buonaccorsi VP, McDowell JR, Graves JE (2001). Reconciling patterns of inter-ocean molecular variance from four classes of molecular markers in blue marlin (Makaira nigricans). Mol Ecol 10: 1179–1196.
Cano JM, Mäkinen HS, Merilä J (2008). Genetic evidence for male-biased dispersal in the three-spined stickleback (Gasterosteus aculeatus). Mol Ecol 17: 3234–3242.
Carreras-Carbonell J, Macpherson E, Pascual M (2006). Population structure within and between subspecies of the Mediterranean triplefin fish Tripterygion delaisi revealed by highly polymorphic microsatellite loci. Mol Ecol 15: 3527–3539.
Cavalli-Sforza LL, Edwards AWF (1967). Phylogenetic analysis models and estimation procedures. Am J Hum Genet 19: 233–257.
Chistiakov DA, Hellemans B, Volckaert FAM (2006). Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255: 1–29.
Ciofi C, Beaumont MA, Swingland IR, Bruford MW (1999). Genetic divergence and units for conservation in the Komodo dragon Varanus komodoensis. Proc R Soc Lond B Biol Sci 266: 2269–2274.
Clarke KR (1993). Nonparametric multivariate analyses of changes in community structure. Aust J Ecol 18: 117–143.
Consuegra S, de Leaniz CG (2007). Fluctuating sex ratios, but no sex-biased dispersal, in a promiscuous fish. Evol Ecol 21: 229–245.
De Innocentiis S, Lesti A, Livi S, Rossi AR, Crosetti D, Sola L (2004). Microsatellite markers reveal population structure in gilthead sea bream Sparus auratus from the Atlantic Ocean and Mediterranean Sea. Fish Sci 70: 852–859.
De Innocentiis S, Sola L, Cataudella S, Bentzen P (2001). Allozyme and microsatellite loci provide discordant estimates of population differentiation in the endangered dusky grouper (Epinephelus marginatus) within the Mediterranean Sea. Mol Ecol 10: 2163–2175.
Ehrenberg SZ, Hansson S, Elmgren R (2005). Sublittoral abundance and food consumption of Baltic gobies. J Fish Biol 67: 1083–1093.
England PR, Cornuet JM, Berthier P, Tallmon DA, Luikart G (2006). Estimating effective population size from linkage disequilibrium: severe bias in small samples. Conserv Genet 7: 303–308.
Estoup A, Jarne P, Cornuet JM (2002). Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11: 1591–1604.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Ewens WJ (1972). Sampling theory of selectively neutral alleles. Theor Popul Biol 3: 87.
Foll M, Gaggiotti O (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993.
Gaggiotti OE, Lange O, Rassmann K, Gliddon C (1999). A comparison of two indirect methods for estimating average levels of gene flow using microsatellite data. Mol Ecol 8: 1513–1520.
Goldstein DB, Linares AR, Cavalli-sforza LL, Feldman MW (1995). An evaluation of genetic distances for use with microsatellite loci. Genetics 139: 463–471.
Goudet J (2001). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.), http://www.unil.ch/izea/softwares/fstat.html.
Goudet J, Perrin N, Waser P (2002). Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol 11: 1103–1114.
Gower JC (1975). Generalized Procrustes Analysis. Psychometrika 40: 33–51.
Gysels ES (2003). Genetic structure and evolutionary patterns of Pomatoschistus gobies of the European continental shelf and in the Mediterranean Sea. PhD-thesis, Katholieke Universiteit Leuven: Leuven.
Gysels ES, Hellemans B, Patarnello T, Volckaert FAM (2004a). Current and historic gene flow of the sand goby Pomatoschistus minutus on the European Continental Shelf and in the Mediterranean Sea. Biol J Linn Soc 83: 561–576.
Gysels ES, Leentjes V, Volckaert FAM (2004b). Small-scale clinal variation, genetic diversity and environmental heterogeneity in the marine gobies Pomatoschistus minutus and P. lozanoi (Gobiidae, Teleostei). Heredity 93: 208–214.
Hamerlynck O (1990). The identification of Pomatoschistus minutus (Pallas) and Pomatoschistus lozanoi (de Buen) (Pisces, Gobiidae). J Fish Biol 37: 723–728.
Hardy OJ, Charbonnel N, Freville H, Heuertz M (2003). Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482.
Hardy OJ, Vekemans X (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hauser L, Carvalho GR (2008). Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish and Fisheries 9: 333–362.
Hedrick PW (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Hewitt G (2000). The genetic legacy of the quaternary ice ages. Nature 405: 907–913.
Huyse T, Van Houdt J, Volckaert FAM (2004). Paleoclimatic history and vicariant speciation in the ‘sand goby’ group (Gobiidae, Teleostei). Mol Phylogenet Evol 32: 324–336.
Jackson DA (1995). Protest—a Procrustean randomization test of community environment concordance. Ecoscience 2: 297–303.
Johannesson K, André C (2006). Life on the margin: genetic isolation and diversity loss in a peripheral marine ecosystem, the Baltic Sea. Mol Ecol 15: 2013–2029.
Jones AG, Walker D, Kvarnemo C, Lindström K, Avise JC (2001). How cuckoldry can decrease the opportunity for sexual selection: Data and theory from a genetic parentage analysis of the sand goby, Pomatoschistus minutus. Proc Natl Acad Sci USA 98: 9151–9156.
Jørgensen HBH, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V (2005). Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol Ecol 14: 3219–3234.
Koskinen MT, Hirvonen H, Landry PA, Primmer CR (2004). The benefits of increasing the number of microsatellites utilized in genetic population studies: an empirical perspective. Hereditas 141: 61–67.
Larmuseau MHD, Guelinckx J, Hellemans B, Van Houdt JKJ, Volckaert FAM (2008). Fast PCR-RFLP method facilitates identification of Pomatoschistus species in the North Atlantic. J Appl Ichthyol 24: 342–344.
Larmuseau MHD, Hellemans B, Van Houdt JKJ, Volckaert FAM (2007). Development and characterization of nine polymorphic microsatellite markers in the sand goby Pomatoschistus minutus (Gobiidae). Mol Ecol Notes 7: 147–149.
Larmuseau MHD, Raeymaekers JAM, Ruddick KG, Van Houdt JKJ, Volckaert FAM (2009a). To see in different seas: spatial variation in the rhodopsin gene of the sand goby (Pomatoschistus minutus). Mol Ecol 18: 4227–4239.
Larmuseau MHD, Van Houdt JKJ, Guelinckx J, Hellemans B, Volckaert FAM (2009b). Distributional and demographic consequences of Pleistocene climate fluctuations for a marine demersal fish in the north-eastern Atlantic. J Biogeogr 36: 1138–1151.
Lindström K, St Mary C, Pampoulie C (2006). Sexual selection for male parental care in the sand goby, Pomatoschistus minutus. Behav Ecol Sociobiol 60: 46–51.
Luikart G, Cornuet JM (1999). Estimating the effective number of breeders from heterozygote excess in progeny. Genetics 151: 1211–1216.
Lukoschek V, Waycott M, Keogh JS (2008). Relative information content of polymorphic microsatellites and mitochondrial DNA for inferring dispersal and population genetic structure in the olive sea snake, Aipysurus laevis. Mol Ecol 17: 3062–3077.
Maes J, Stevens M, Ollevier F (2005). The composition and community structure of the ichthyofauna of the upper Scheldt estuary: synthesis of a 10-year data collection (1991–2001). J Appl Ichthyol 21: 86–93.
Mantel N (1967). The detection of disease clustering and a generalised regression approach. Cancer Res 27: 209–220.
Meirmans PG (2006). Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60: 2399–2402.
Miller PJ (1986). Gobiidae. In: Whitehead PJP, Bauchot M-L, Hureau JC, Nielsen J and Tortononese E (eds). Fishes of the north-eastern Atlantic and Mediterranean. UNESCO: Paris, pp 1019–1085.
Nesbø CL, Rueness EK, Iversen SA, Skagen DW, Jakobsen KS (2000). Phylogeography and population history of Atlantic mackerel (Scomber scombrus L.): a genealogical approach reveals genetic structuring among the eastern Atlantic stocks. Proc R Soc Lond B Biol Sci 267: 281–292.
O’Reilly PT, Canino MF, Bailey KM, Bentzen P (2004). Inverse relationship between FST and microsatellite polymorphism in the marine fish, walleye pollock (Theragra chalcogramma): implications for resolving weak population structure. Mol Ecol 13: 1799–1814.
Oksanen J, Kindt R,, Legendre P, O’Hara RB (2007). Community Ecology Package version 1.8–5: vegan, http://cran.r-project.org/.
Pampoulie C, Gysels ES, Maes GE, Hellemans B, Leentjes V, Jones AG et al (2004). Evidence for fine-scale genetic structure and estuarine colonisation in a potential high gene flow marine goby (Pomatoschistus minutus). Heredity 92: 434–445.
Pasquaud S, Girardin M, Elie P (2004). Diet of gobies of the genus Pomatoschistus (P. microps and P. minutus), in the Gironde estuary (France). Cybium 28: 99–106.
Peel D, Ovenden JR, Peel SL (2004). NeEstimator: software for estimating effective population size, Version 1.3. Queensland Government; Department of Primary Industries and Fisheries, http://www.dpi.qld.gov.au/cps/rde/dpi/hs.xsl/28_6908_ENA_HTML.htm.
Peijnenburg KTCA, Fauvelot C, Breeuwer AJ, Menken SBJ (2006). Spatial and temporal genetic structure of the planktonic Sagitta setosa (Chaetognatha) in European seas as revealed by mitochondrial and nuclear DNA markers. Mol Ecol 15: 3319–3338.
Poulsen NA, Nielsen EE, Schierup MH, Loeschcke V, Gronkjaer P (2006). Long-term stability and effective population size in North Sea and Baltic Sea cod (Gadus morhua). Mol Ecol 15: 321–331.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Queller DC, Goodnight KF (1989). Estimating relatedness using genetic markers. Evolution 43: 258–275.
Raymond M, Rousset F (1995). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rodrigues P, Reis-Henriques MA, Campos J, Santos MM (2006). Urogenital papilla feminization in male Pomatoschistus minutus from two estuaries in northwestern Iberian Peninsula. Mar Environ Res 62: S258–S262.
Ruzzante DE, Taggart CT, Cook D (1998). A nuclear DNA basis for shelf- and bank-scale population structure in northwest Atlantic cod (Gadus morhua): Labrador to Georges Bank. Mol Ecol 7: 1663–1680.
Shaw PW, Arkhipkin AI, Al-Khairulla H (2004). Genetic structuring of Patagonian toothfish populations in the Southwest Atlantic Ocean: the effect of the Antarctic Polar Front and deep-water troughs as barriers to genetic exchange. Mol Ecol 13: 3293–3303.
Slatkin M (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Smouse PE, Long JC, Sokal RR (1986). Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool 35: 627–632.
Stefanni S, Gysels ES, Volckaert FAM, Miller PJ (2003). Allozyme variation and genetic divergence in the sand goby, Pomatoschistus minutus (Teleostei: Gobiidae). J Mar Biol Ass UK 83: 1143–1149.
Tamura K, Nei M (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526.
Verhoeven KJF, Simonsen KL, McIntyre LM (2005). Implementing false discovery rate control: increasing your power. Oikos 108: 643–647.
Wang JL, Whitlock MC (2003). Estimating effective population size and migration rates from genetic samples over space and time. Genetics 163: 429–446.
Waples RS (1989). A generalized-approach for estimating effective population-size from temporal changes in allele frequency. Genetics 121: 379–391.
Waples RS (2006). A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7: 167–184.
Waples RS, Do C (2008). LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resources 8: 753–756.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Acknowledgements
We thank the following persons who kindly provided the samples used in this study: Pilar Drake (CSIC, Spain), Maria Järvi-Laturi (University of Helsinki, Finland), Kai Lindström (Åbo Akademi University, Finland), Lotta Kvarnemo (Göteborgs Universitet, Sweden), Ian McCarthy (University of Wales, Bangor, UK), Mario Lepage (Ifremer, France), Jef Guelinckx and Alain Crivelli (Station Biologique de la Tour du Valat, France). We also thank three anonymous referees, Ronny Decorte, Luisa Orsini, Hendrik Larmuseau, Gregory Maes, Wendy Wiertz and Dirk Schaerlaekens for interesting discussions, and Dorien Daneels for the simulation analyses. The first author received a PhD fellowship of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen). This study is part of the WESTBANKS project financed by the Belgian Federal Office for Scientific, Technical and Cultural Affairs (contract no. SD/BN/01A).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Larmuseau, M., Raeymaekers, J., Hellemans, B. et al. Mito-nuclear discordance in the degree of population differentiation in a marine goby. Heredity 105, 532–542 (2010). https://doi.org/10.1038/hdy.2010.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.9
Keywords
This article is cited by
-
Shedding light on variation in reproductive success through studies of population genetic structure in a Southeast Pacific Coast mussel
Heredity (2023)
-
Reviewing the genetic evidence for sex-biased dispersal in elasmobranchs
Reviews in Fish Biology and Fisheries (2021)
-
Genomic and phenotypic consequences of two independent secondary contact zones between allopatric lineages of the anadromous ice goby Leucopsarion petersii
Heredity (2020)
-
Phylogeography and genetics of the globally invasive snail Physa acuta Draparnaud 1805, and its potential to serve as an intermediate host to larval digenetic trematodes
BMC Evolutionary Biology (2018)
-
Genetic homogeneity of the invasive lionfish across the Northwestern Atlantic and the Gulf of Mexico based on Single Nucleotide Polymorphisms
Scientific Reports (2018)
