
Abstract
In pentaploid dogroses, Rosa section Caninae (2n=5x=35), the pollen transmits one basic genome (x=7) derived from the seven segregating bivalents, whereas the egg transmits four basic genomes (4x=28) one set derived from the segregation of seven bivalents and three sets of univalent-forming chromosomes. Chromosomes from all five genomes carry 18-5.8-26S nuclear ribosomal DNA (rDNA) sites. This mode of sexual reproduction, known as permanent odd polyploidy, can potentially lead to the independent evolution of rDNA on bivalent- and univalent-forming chromosomes. To test this hypothesis, we analyzed rRNA gene families in pollen and somatic leaf tissue of R. canina, R. rubiginosa and R. dumalis. Six major rRNA gene families (α, β, β′ γ, δ and ɛ) were identified based on several highly polymorphic sites in the internal transcribed spacers (ITSs). At least two of the major rRNA gene families were found in each species indicating that rDNAs have not been homogenized across subgenomes. A comparison of ITS1 sequences from leaf and pollen showed differences: the shared β rRNA gene family was more abundant among pollen clones compared to leaf clones and must constitute a major part of the rDNA loci on bivalent-forming chromosomes. The γ and δ families were underrepresented in pollen genomes and are probably located predominantly (or solely) on the univalents. The results support the hypothesis that pentaploid dogroses inherited a bivalent-forming genome from a common proto-canina ancestor, a likely donor of the β rDNA family. Allopolyploidy with distantly related species is likely to have driven evolution of Rosa section Caninae.
Similar content being viewed by others


Introduction
Within the genus Rosa L., the section Caninae DC forms a large and well-defined group of polyploid taxa (x=7) known as dogroses. Pentaploids are most common but tetraploids and hexaploids also occur (for review, see Wissemann, 2003). All dogrose species have the peculiar ‘canina meiosis’ described more than 80 years ago (Täckholm, 1920). In pollen meiosis, only seven bivalents are formed, the other chromosomes remain as univalents and are excluded from pollen. Thus, pollen only carries seven chromosomes derived from the segregation of the bivalent-forming chromosomes. In contrast, all the univalents and seven chromosomes from the segregation of the bivalents are included in viable egg cells, which carry 21, 28 or 35 chromosomes depending on the ploidy level of the parent plant. This unusual meiosis can potentially lead to different patterns of evolution of bivalent- and univalent-forming chromosomes if the same chromosomes are always involved in bivalent formation at each meiosis, as we suspect from karyotype analysis (Lim et al., 2005). Certainly, the transmission of microsatellite and random amplification of polymorphic DNA (RAPD) markers in interspecific crosses are consistent with that hypothesis (Werlemark and Nybom, 2001; Wissemann, 2002; Nybom et al., 2004, 2006). The absence of extensive pairing between the four genomes inherited through the egg cell was demonstrated in a cytogenetic study of a gynogenetic haploid of R. canina (obtained after pollination with γ-irradiated pollen), in which chromosomes at meiosis I in pollen mother cells remained predominantly unpaired (Lim et al., 2005).
In most eukaryotes 5S and 18-5.8-26S ribosomal DNA (rDNA) units occur in tandem arrays at one (as in species of section Caninae, Lim et al., 2005) or several rDNA loci per basic genome. Each large rDNA unit contains the 18S, 5.8S and 26S rDNA subunits, the internal transcribed spacers (ITSs) sequences and the intergenic spacer (IGS). The genes are highly conserved even between eukaryotes and prokaryotes, whereas divergence of ITS is sufficient to resolve species relationships within most genera (for reviews see Alvarez and Wendel, 2003; Nieto Feliner and Rossello, 2007). Of particular interest to evolutionary biologists is the pattern of divergence of the whole rDNA array, which is often influenced by sequence homogenization that functions to replace existing genic units with variants of that unit over time, a process known as concerted evolution (Dover, 1982; Eickbush and Eickbush, 2007).
The Caninae species, with their unusual asymmetric meiosis, present a unique opportunity to study evolution of repeated sequences with and without chiasmatic recombination at meiosis. In this paper, we asked the following questions: (i) Is there evidence for rDNA homogenization in pentaploid dogroses? (ii) Has concerted evolution homogenized rDNA arrays across all or some genomes? (iii) What is the distribution of rRNA gene families between bivalent and univalent-forming genomes? (iv) Are ITSs on univalent-forming chromosomes less homogenous than those on bivalent-forming chromosomes? To address these questions, we used southern blot hybridization and extensive cloning of ITS repeats in three pentaploid dogroses; R. canina, R. rubiginosa and R. dumalis. We also compared ITS sequence profiles from leaf DNA (containing complete chromosomal sets) and pollen grains thought to contain mainly bivalent-forming genetic material.
Materials and methods
Plant material
Rosa canina (accessions 1051 and 1073), R. rubiginosa (1408 and 0391), R. dumalis (8701), R. sherardii (1402) and R. caesia (504) plants were collected as root suckers in the wild and subsequently grown at Balsgård, South Sweden. Each plant has previously been shown to be pentaploid (2n=5x=35) using flow cytometry (Nybom et al., 2004, 2006). The pollen contains both haploid gametophytic tissue (pollen nuclei) and probably some traces of diploid sporophytic tissue (for example, anther walls). In comparison to the leaf-derived DNA, pollen-derived DNA should, however, contain a higher amount of bivalent-forming genetic material and a lower amount of univalent-forming material.
Isolation of genomic DNA
Leaf DNA was isolated according to a standard cetyl trimethylammonium bromide (CTAB) method (Saghai-Maroof et al., 1984) following minor modifications described in Fojtova et al. (1998). The pollen grains were collected in dry weather to avoid absorption of moisture and kept frozen (−20 °C) until use. For isolation of genomic DNA we used ∼1 g of mature pollen, equivalent to about 10 000 pollen grains, assuming the dry grain weight to be 0.13 mg per pollen grain. The tissue was homogenized in a modified CTAB buffer (1.4% w/v cetylammonium bromide, 0.07 M Tris-HCl, pH 8.0, 1.14 M NaCl and 0.1% 2-mercaptoethanol) with a micropestle (Eppendorf) in a 1.5 ml tube. After the isopropanol precipitation, the crude DNA was treated with RNase-A (40 μg ml−1) for 10 min at 37 °C, then with Protease-K (100 μg ml−1) for 2 h at 50 °C. Enzymatic treatments were terminated with phenol/chloroform extraction and ethanol precipitation. The final product was dissolved in 10 μl of 10mM Tris-HCl, pH 8.0 1mM EDTA (TE) buffer. DNA concentration was estimated at optical density (OD) 260 nm using a Nanodrop spectrophotometer. The total yield varied from 1 to 6 ng DNA.
Southern blot hybridization
For southern blot hybridization, purified DNAs were digested with an excess of restriction enzymes (10 U/μg DNA), separated by gel electrophoresis, and blotted onto nylon membranes (Hybond XL, GE Healthcare; http://www.gehealthcare.com). We used methylation-insensitive restriction enzymes (RsaI, HinfI, MboI, TaqI and BstNI) to avoid inhibition of enzyme activity due to cytosine methylation. The southern blot hybridization was carried out in a modified Church–Gilbert buffer (Lim et al., 2000) using the ITS1 rDNA labeled with [32P] dCTP (ICN). The ITS1 probe was a cloned ∼700 bp fragment from R. rubiginosa (accession number EU168708) containing 5′-18S (part)-ITS1-5.8S (part)-3′. The hybridization signals were measured using a PhosphorImager (Storm, GE Healthcare; http://www.gehealthcare.com), and data processed by ImageQuant software.
Cloning analysis
Internal transcribed spacers sequences were obtained by PCR amplification of genomic DNA. In a 25 μl reaction, we used 0.1–1 ng genomic DNA as template, 4 pmol of each primer, 2.4 nmol of each dNTP and 0.4 units of DyNAzyme II DNA polymerase (Finnzymes, Espoo, Finland). Cycling conditions were as follows: initial denaturation step (94 °C, 180 s) and 15–35 cycles (94 °C, 20 s; 57 °C, 30 s and 72 °C, 30 s) with 18S-FOR and 5.8S-REV primers. The primer sequences for ITS1 were as follows: 18S-FOR: 5′-GCGCTACACTGATGTATTCAACGAG-3′ and 5.8S-REV: 5′-CGCAACTTGCGTTCAAAGACTCGA-3′ (Kovarik et al., 2005). The PCR products were purified using a Qiagen column and cloned into a dT vector (pDrive, Qiagen, http://www.qiagen.com/). We also considered the possibility of differential amplification of GC-rich and GC-poor templates (Keller et al., 2006) and included 5% dimethyl sulfoxide (DMSO) in a PCR reaction. We found that DMSO had no effect on the proportion of amplified gene families. Sequencing was carried out by the dideoxy chain-termination method at the BioTEch Vienna, Austria using the 5.8S-REV sequencing primer. The ITS1-5.8S-ITS2 sequences were amplified using 18S-FOR and 26S-REV (5′-CTTTTCCTCCGCTTATTGATATGC-3′) primers (Kovarik et al., 2005) yielding ∼1.0-kb products in all species.
Genomic-cleaved amplified restriction polymorphism was used to obtain information on the entire population of PCR products. About 0.5 ng of genomic DNA was used as a template in the PCR. The products were digested with restriction enzymes and subjected to electrophoresis in a 2% agarose gel using methods used in Matyasek et al. (2007).
Sequence alignment and analysis of nucleotide diversity
The data were assembled in the window of the Wisconsin GCG package software (version 10.3. Accelrys Inc., San Diego, CA, USA). Alignments were carried out using the PILE UP (implemented within the GCG) or the CLUSTAL W at The European Bioinformatics Institute (EMBL-EBI) server. Bootstrap analysis using 500 repetitions was carried out using the PHYLOWIN program (Galtier et al., 1996). The topology of the phylogenic tree was interpreted as by the following categories of bootstrap support: unsupported (<60%), weakly supported (60–80%) and strongly supported (80–100%). The genetic distances between individual clones were calculated from a table of pairwise similarities of aligned sequences (OldDistances, GCG package, Accelrys, San Diego, CA, USA). In total, the ITS data sets comprised ∼200 sequences from three species. The aligned sequences were inspected by eye, and gene families identified based on the polymorphic sites threshold (>5%). Gene family frequencies were analyzed using a χ2-test to determine whether the pollen rRNA gene families differed significantly with those obtained from leaf DNA. Representative sequences of each ITS family were deposited in the EMBL/GenBank under the accession numbers EU168707-EU168711 (ITS1 clones). The ITS1-5.8S-ITS2 clones were deposited under nos. FM164946-FM164950 and FM164423-FM164424.
Fluorescent in situ hybridization
Methods followed those described in Lim et al. (2005). Briefly, the clone pTa71, containing the 18-5.8-26S rDNA unit from Triticum aestivum was labeled with digoxigenin-11-dUTP and hybridized in situ to chromosomes to give an estimated ⩾85% sequence identity. Sites of probe hybridization were detected with fluorescein (FITC)-conjugated antidigoxigenin (Roche Biochemicals, http://www.roche.com). Chromosomes were counterstained with DAPI (4′,6-diamidino-2-phenylindole).
Results
Southern blot analysis of rRNA gene families
To study the occurrence and degree of rDNA homogenization, we used southern blot hybridization of restricted DNA and an ITS1 sequence from R. rubiginosa as a probe. Out of several enzymes tested, only BstNI gave restriction fragment length polymorphism profiles that differed between R.canina, R. rubiginosa, R. dumalis, R. sherardii and R. caesia (Figure 1). In all species, the probe hybridized to a common ∼500-bp BstNI-band possibly representing a highly conserved rDNA family. In addition, the probe hybridized to several bands in a species-specific manner. For example, a high-molecular-weight band was found in R. canina (weak) and R. rubiginosa (strong), but not in other species. Three species, R. dumalis, R. sherardii and R. caesia, shared a doublet of closely migrating bands. There was no, or minimal, differences in hybridization profiles between the two accessions from each of R. canina and R. rubiginosa. The blot hybridization with DNAs digested with other enzymes (Supplementary Figure S1) revealed simple profiles of distinct bands indicating a high homogeneity of dogrose DNAs at restriction sites studied.

Restriction site polymorphisms in rDNA among species of section Caninae detected by southern blot hybridization. The genomic DNAs were digested with BstNI and hybridized against the ITS1 probe. r+c, bands specific for R. rubiginosa and R. canina; d+s+cs, bands specific for R. dumalis, R. sherardii and R. caesia; common bands to all species are indicated. ITS, internal transcribed spacers.
Cloning of ITS from somatic leaf genomes
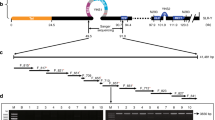
We examined sequence polymorphisms in ITS1 in R. canina, R. rubiginosa and R. dumalis (>20 ITS1 sequences per species). ITS1 sequences of dogroses are 262±1 bp long (Figure 2). The length variation was caused by variable numbers of nucleotides in a G-tract close to the 18S subunit. Cloning experiments yielded 94 sequences comprising 24 628 nucleotides. Total alignment matrix revealed 40 polymorphic sites (∼15%) reflecting different ribotypes (32) in Canina genomes (Figure 3a, Supplementary Figure S2). Most ribotypes were low copy number (present in one or few clones). The meaningful interpretation of these low-frequency polymorphisms would be a mutation noise and polymerase/sequencing errors. However, there were several high-copy polymorphisms (>5% of total ITS1 clones) arising from nine mutation hotspots (Figure 2, Table 1). These hotspots (diagnostic sites) were used to identify major rRNA gene families of Caninae (Figure 3b, Table 2). Seven diagnostic sites overlapped with those previously determined in a genus level study of ITS1 sequences (Ritz et al., 2005), while another five polymorphic sites detected in that study were not identified as polymorphic in our data sets. Substitutions at diagnostic sites involved both transitions (71%) and transversions (29%), percentages that are similar to those reported previously in other species (Buckler et al., 1997; Zhang et al., 2002). Most mutations involved C → T and G → A transitions and possibly originated from deamination of methylcytosines. Four out of five transitions occurred in CG dinucleotides known to be a primary target of cytosine methylation in plant genomes (Kovarik et al., 1997). Two mutation hotspots were located to polymorphic dinucleotide motifs at positions −111/−112 and −137/−138. At these hotspots, the mutation of a first nucleotide was often compensated/associated with a mutation of a nucleotide in the second position possibly maintaining a fixed secondary structure of the primary rRNA transcript (computer models based on RNA thermal stability are shown in Supplementary Figure S3).

Consensus sequence of ITS1-5.8S-ITS2 in R. canina, R. rubiginosa and R. dumalis. Positions are numbered with respect to the first nucleotide of the 5.8S gene (based on the annotated Nicotiana sequences taken from Genebank). The mutation hot spots are indicated with gray shading. The sequences of genic regions are in italics. (∼) indicates sites of frequent nucleotide insertions/deletions. Keys for variable nucleotides (IUPAC): K=T,G; R=A,G; Y=T,C ITS, internal transcribed spacers.

Frequency of ITS1 ribotypes within the 94 leaf clones from three Rosa species (a). The full list of polymorphisms is given in Supplementary Figure S2. The categorization of most common ribotypes (b). The gene families were defined based on consensus sequences (Table 2) of ribotypes occurring at >5% frequency. ITS, internal transcribed spacers.
Phylogenetic relationships between the six major families are shown in Figure 4. One well-supported clade comprised the α, β and β′ rDNA families. The members of this relatively GC-rich (59%) clade differ from each other by 1–2 substitutions at the diagnostic sites (Table 2). Both α and β(β′) families differed by two substitutions at positions −111/−112. The β′ subfamily differed from β by the presence of a unique G → A mutation at −102. The second clade comprised three families, each having a lower GC content (55–56%). Embedded within this clade, the γ and δ families form a subclade, with both families characterized by a single substitution at position −56. The ɛ family branch was less stable and the sequence (Table 2) suggests that it was derived through recombination between the two primary clades. Currently, it is not known whether the ɛ family is a true endogenous recombinant or whether it arose during PCR reaction as an artifact (Cronn et al. 2002). Overall divergence within clones of the same family was low (0–0.003), while variation across the families was higher (0.002–0.025). We also included published ITS1 sequences from the database in the alignment. It is evident that the sequences from nondogrose species, R. gallica and R. wichurana, did not form separate branches but were instead embedded within the γ, δ and ɛ clades. This pattern is consistent with the hypothesis that some ITS families are shared between both dogrose and nondogrose species. In contrast, the R. canina ITS1 family (accession number AJ631923), previously found to be present in all investigated dogroses, but not in any other Rosa species (Ritz et al., 2005), fell within the β clade. Maximum parsimony analysis (Kimura, 1980) revealed that ITS1 sequences from the different species were frequently intermingled, although each clade was typically populated predominantly with sequences from one species (Supplementary Figure S4). Thus, most gene families were shared between the species, while some were species-specific (Table 3), consistent with the southern hybridization profiles. The inclusion of Rubus ITS1 within the tree as an out group resulted in a near loss of resolution of Caninae clades probably due to high divergence of Rubus and Rosa sequences (>0.1).

Phylogenetic relationships between ITS1 families. The tree has been constructed based on the alignment of consensus family sequences using a neighbor joining method (Jukes–Cantor distance model). Statistical analysis was carried out using the PHYLOWIN program allowing for 500 repetitions. Bootstrap values are indicated. The published ITS1 sequences from nucleotide database are annotated by the accession numbers. ITS, internal transcribed spacers.
We further extended the analysis for ITS2 and amplified ∼1.0 kb 18S-26S fragment comprising ITS1-5.8S-ITS2 sequences and sequenced five clones from each species. The average GC content of ITS2 was 58%. ITS2 was generally less polymorphic than ITS1; there were three indels, while there were no conserved substitutions found. It appeared that members of α and β families had ITS2 of 208 bp, while the members of γ and ɛ families had the ITS2 of 209 bp (Supplementary Figure S5).
Distribution of ITS1 families in pollen genomes
Pollen grains are thought to contain an increased proportion of chromosome sets derived from the segregation of bivalents only. Comparison of the occurrence and abundance of rDNA families in pollen and in somatic tissues, respectively, has the potential to reveal overall differences in rDNA families occurring on bivalent- and univalent-forming chromosomes. We, therefore, isolated genomic DNA from pollen and cloned ITS1 subregions from three dogrose species: two accessions of R. canina and R. rubiginosa and one accession of R. dumalis. These data were compared with sequences from leaf material of the same accessions (Tables 1 and 3). Two categories were compared with χ2 analysis, clones belonging to the β-family versus clones belonging to any of the other families. When calculated separately for each accession, results were not significant for any of the R. canina accessions (accession 1051, χ2=0.8, NS (nonsignificant), and accession 1073, χ2=2.5, NS). By contrast, significant values were obtained for R. dumalis, χ2=3.8, P=0.05 and for the two R. rubiginosa accessions (accession 0931, χ2=4.9, P<0.05 and accession 1408, χ2=5.2, P<0.025). Finally, an analysis was conducted with clones pooled across all five accessions, χ2=10.7, P<0.01. In all of these analyses, even those yielding nonsignificant χ2 values, the β clones were most common in the pollen DNA, whereas clones from other families were more common in the leaf DNA.
The limited numbers of sequenced clones (∼20 per accession) preclude a definite interpretation of our results, as there are probably thousands of copies of rDNA units in each basic genome. We, therefore, carried out genomic-cleaved amplified restriction polymorphism analysis to obtain a broad overview of rDNA types in the genome. Digestion of R. rubiginosa PCR products with BstNI generated a larger fraction of lower molecular weight fragments in pollen than leaf genomic DNA (Figure 5). As the β family (but not γ) contains a conserved BstNI site, we conclude that the β family was overrepresented in pollen DNA, in agreement with sequence analysis. In R. canina, cleavage of leaf ITS1 PCR product yielded four TaqI and TaiI fragments while there were only three in pollen DNA. Thus, leaf DNA seems to contain extra rRNA gene family(s) not found in pollen. Attempts to analyze pollen DNA by southern blot hybridization were unsuccessful due to the difficulties in extracting large amounts of DNA from pollen.

g-CAPS analysis of ITS1 in R. rubiginosa and R. canina. The enzymes used for the detection of sequence polymorphisms are given. Left panel (R. rubiginosa): the ratio of the undigested (700 bp) and digested (480 bp and 220 bp) BstNI fragments is given below the lanes. Right panel (R. canina): restriction fragments occurring specifically in leaf material are indicated by asterisks. g-CAPS, genomic-cleaved amplified restriction polymorphism; ITS, internal transcribed spacers.
rDNA chromosomal distributions
There are five 18-5.8-26S. rDNA sites in pentaploid R. canina, one per basic genome (Lim et al., 2005). Three of these chromosomes have similar-sized rDNA loci and secondary constrictions, two of which are thought to pair at meiosis (Lim et al., 2005). A fourth locus also has a secondary constriction and is the largest rDNA locus. A fifth locus carries no secondary constriction and is probably silent (Figures 6a and b). In early pollen grain meiosis (leptotene), the rDNA loci are all associated, frequently visible as two oppressed bundles of arrays (Figure 5c).

Cell spreads of R. canina probed with pTa71 for 18-5.8-26S rDNA (digoxigenin-labeled, FITC detected, yellow fluorescence). (a) Root tip mitotic cell labeled for rDNA and (b) superimposed with DAPI signal (blue) for chromatin. Note the five rDNA sites, small arrowheads indicate decondensed or partially decondensed rDNA loci, and the large arrowhead indicates a condensed, probably inactive site. The inactive locus occurs in a hemizygous configuration and probably locates to a univalent chromosome. (c) Pollen grain meiotic cell at leptotene showing the association of rDNA loci (arrow). Scale bar=10 μm. rDNA, ribosomal DNA.
Discussion
rDNA families and genome composition of dogroses
On the basis of the transmission of microsatellite and RAPD markers in interspecific crosses, we have recently proposed tentative genomic compositions for five dogrose species (Nybom et al., 2006). In all of these species, there was one homogenous diploid genome, evidently taking part in bivalent formation and meiotic recombination, and two (in tetraploids) or three (in pentaploids) univalent genomes with variable levels of similarity to one another and to the bivalent-forming genomes. The presence of at least two diverged genomes in each species is supported by our ITS1 data as there are at least two diverged ITS1 families in each species. The β family occurring in all species is virtually identical to the C-type family previously identified by Ritz et al. (2005). As this C-type family occurs in all dogrose species so far investigated but not in any other taxa, it has been hypothesized to represent a proto-canina type rDNA. In contrast, the γ and δ families are homologous to ITS1 types found in R. gallica and R. wichurana (Figure 4). These data support the hypothesis that the dogroses are allopolyploids that arose by repeated interspecies hybridization of a common ancestral proto-canina genome (a donor of the β rDNA family) with donors of diverged γ and δ families. R. gallica belongs to section Rosa, while R. wichurana belongs to section Synstylae, both of which have been shown to have considerable affinities with section Caninae when investigated with amplified fragment length polymorphism (AFLP) markers (Koopman et al., 2008). Probably, species within these two sections may have been the donors of haploid genome(s) in R. rubiginosa and R. dumalis. The β ITS1 family differed from the γ family by six nucleotide substitutions within the 262 bp of ITS1. Given that the published substitution rates are 8.34 × 10−9 –3.89 × 10−8 per site and year for ITS (Zhang et al., 2001; Weiss-Schneeweiss et al., 2007) it follows that the β and γ families have separated 9–28 Myr ago. In contrast, dogroses are often believed to be considerably more recent, deriving from postglacial allopolyploidization events (Wissemann, 2006). If so, the origin of the rDNA families must predate the Caninae divergence.
Absence of extensive interlocus rDNA homogenization
Ribosomal RNA genes, as other tandemly arranged repeats, can evolve through the concerted evolution that keeps the arrays more homogenous than would be expected by chance. As a consequence, in many allopolyploid species, parental rDNA arrays have been homogenized toward one parental type, a process called interlocus homogenization. Most studies reveal extensive interlocus homogenization even in recently formed allopolyploid species (Franzke and Mummenhoff, 1999; Kovarik et al., 2005 and for review see Volkov et al., 2007), although the presence of multiple rDNA families has been observed and reported previously (for example, Buckler et al., 1997; Harpke and Peterson, 2006; Keller et al., 2006). Of the species analyzed, R. rubiginosa had the most homogenous spectrum of rDNA types involving two highly diverged ITS1 families (β and γ). A low level of variation of microsatellite markers was also observed in R. rubiginosa, resulting in the proposal of a genomic composition AAA1BB1 (Nybom et al., 2006). In contrast, R. dumalis had clones from five different ITS1 families (α, β, β′, γ and δ). Similarly, there was a high microsatellite diversity in this species with a proposed genomic composition AABCD (Nybom et al., 2006). The presence of section Caninae in species of multiple ITS families, and of their different distributions in pollen and leaf materials, indicates that the interlocus homogenization has not gone to completion/fixation. There are several explanations for this. First, intergenomic recombination leading to interlocus homogenization might be compromised in univalent-forming chromosomes, perhaps because the rDNA locus association at leptotene (Figure 6c) is lost during the formation of bivalents preventing meiotic recombination. The nucleolar organizer regions (NORs) on univalent-forming chromosomes might be behaving like individual B-chromosomes of Crepis that apparently do not recombine with NORs on autosomes (Leach et al., 2005). Secondly, rDNA units on univalent-forming chromosomes could be epigenetically modified and this might prevent homogenization (Dadejova et al., 2007). Finally, if the polyploids are of recent origin, there may not have been sufficient time to allow for extensive interlocus homogenization.
ITS composition in bivalent- and univalent-forming chromosomes
The different ratios of individual ITS families in pollen and leaf tissues support the hypothesis that the same chromosome sets form univalents or bivalents at each meiosis. For example, in both R. canina accessions, the major α family of ITS sequences was underrepresented in pollen material, while the β family was overrepresented. Similarly, R. rubiginosa pollen had an overabundance of the β family. R. dumalis pollen completely lacked the unique species-specific δ family, while both β and γ families were present.
It is significant that the ubiquitously present β family appears to predominate in pollen irrespective of species. Probably NORs on bivalent-forming chromosomes are preferentially composed of these units. If so, the bivalent-forming chromosomes may be more similar to each other when compared across species than the univalents. This is supported by our previous results showing that the microsatellite alleles in bivalent-forming genomes were shared between genotypes even from widely different taxa, whereas univalent-residing microsatellite alleles closely mirror the level of inter- and intraspecific relatedness (Nybom et al., 2006). Similar observations were made in an older study demonstrating skewed transmission of RAPD markers during interspecies crosses (Werlemark and Nybom, 2001). Given that the β family is likely to occur on bivalent-forming chromosomes (where it is transmitted to both pollen and egg cells) it is likely that all or some of the other families are located on univalent-forming chromosomes transmitted by egg cells only. The occurrence of non-β families in pollen material can be potentially explained by variable amounts of contaminating somatic tissue or immature diploid pollen present in pollen material. Thus, anthers are expected to contain both some sporophytic tissue (anther walls) and pollen cells. In dogroses, the only viable pollen cells contain seven bivalent-derived chromosomes, whereas other pollen cells (containing a variable number of univalents) shrivel up and become nonviable.
Intralocus homogenization influences rDNA arrays on univalent- and bivalent-forming chromosomes
Recombination is proposed as a major mechanism for driving concerted evolution (Eickbush and Eickbush, 2007; Ganley and Kobayashi, 2007). If recombination is driven by meiosis, it may be expected that arrays on bivalent-forming chromosomes will be different from those on univalents. However, comparison of multiple clones of each family failed to show substantial intrafamily heterogeneity for any family, irrespective of their predominant (or potentially absolute) location to univalents or bivalents. For example, in R. canina, calculated nucleotide diversity (Kimura, 1980) between all pairs of sequences was 0.006±0.0007 and 0.007±0.003 for α and β rDNA families, respectively. Similarly, the within-family variability was comparable for both major β and γ rDNA families in R. rubiginosa (average divergence <0.01).
There can be several explanations for homogenization of rDNA within univalents in the apparent absence of meiotic recombination. Firstly, rare meiotic cycles involving ‘univalent’-forming chromosomes could homogenize rDNA units over generations. Variation in ploidy level within some populations (Nybom et al., 2006) may suggest that rare pairing involving univalents actually does take place. Secondly, univalent chromosomes could be of recent origin and hence there might not have been enough time for the accumulation of mutations. Thirdly, concerted evolution may operate on rDNA arrays in the absence of meiotic recombination. In support of the latter hypothesis, transposons on nonrecombining Y chromosomes were shown to be equally homogeneous to those on autosomes in Melandrium (Kejnovsky et al., 2007). In addition, rDNA units on nonrecombining B-chromosomes are relatively homogenous within arrays (intralocus homogenization) but do not show interlocus homogenization (Leach et al., 2005). It would be informative to study epigenetic modification and expression patterns of NORs to see whether NORs on univalent- and bivalent-forming genomes are equally active.
References
Alvarez I, Wendel JF (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol 29: 417–434.
Buckler ES, Ippolito A, Holtsford TP (1997). The evolution of ribosomal DNA: divergent paralogues and phylogenetic implications. Genetics 145: 821–832.
Cronn R, Cedroni M, Haselkorn T, Grover C, Wendel JF (2002). PCR-mediated recombination in amplification products derived from polyploid cotton. Theor Appl Genet 104: 482–489.
Dadejova M, Lim KY, Souckova-Skalicka K, Matyasek R, Grandbastien MA, Leitch A et al. (2007). Transcription activity of rRNA genes correlates with a tendency towards intergenomic homogenization in Nicotiana allotetraploids. New Phytol 174: 658–668.
Dover G (1982). Molecular drive: cohesive mode of species evolution. Nature 9: 111–116.
Eickbush TH, Eickbush DG (2007). Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175: 477–485.
Fojtova M, Kovarik A, Votruba I, Holy A (1998). Evaluation of the impact of S-adenosylhomocysteine metabolic pools on cytosine methylation of the tobacco genome. Eur J Biochem 252: 347–352.
Franzke A, Mummenhoff K (1999). Recent hybrid speciation in Cardamine (Brassicaceae)-conversion of nuclear ribosomal ITS sequences in statu nascendi. Theor Appl Genet 98: 831–834.
Galtier N, Gouy M, Gautier C (1996). SeaView and Phylo_win, two graphic tools for sequence alignment and molecular phylogeny. Comput Applic Biosci 12: 543–548.
Ganley AR, Kobayashi T (2007). Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17: 184–191.
Harpke D, Peterson A (2006). Non-concerted ITS evolution in Mammillaria (Cactaceae). Mol Phylogenet Evol 41: 579–593.
Kejnovsky E, Hobza R, Kubat Z, Widmer A, Marais GA, Vyskot B (2007). High intrachromosomal similarity of retrotransposon long terminal repeats: evidence for homogenization by gene conversion on plant sex chromosomes? Gene 390: 92–97.
Keller I, Chintauan-Marquier IC, Veltsos P, Nichols RA (2006). Ribosomal DNA in the grasshopper Podisma pedestris: escape from concerted evolution. Genetics 174: 863–874.
Kimura M (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120.
Koopman WJM, Wissemann V, De Cock K, Van Huylenbroeck J, De Riek J, Sabatino GJH et al. (2008). AFLP markers as a tool to reconstruct complex relationships: a case study in Rosa (Rosaceae). Am J Bot 95: 353–366.
Kovarik A, Matyasek R, Leitch A, Gazdova B, Fulnecek J, Bezdek M (1997). Variability in CpNpG methylation in higher plant genomes. Gene 204: 25–33.
Kovarik A, Pires JC, Leitch AR, Lim KY, Sherwood AM, Matyasek R et al. (2005). Rapid concerted evolution of nuclear ribosomal DNA in two Tragopogon allopolyploids of recent and recurrent origin. Genetics 169: 931–944.
Leach CR, Houben A, Field B, Pistrick K, Demidov D, Timmis JN (2005). Molecular evidence for transcription of genes on a B chromosome in Crepis capillaris. Genetics 171: 269–278.
Lim KY, Kovarik A, Matyasek R, Bezdek M, Lichtenstein CP, Leitch AR (2000). Gene conversion of ribosomal DNA in Nicotiana tabacum is associated with undermethylated, decondensed and probably active gene units. Chromosoma 109: 161–172.
Lim KY, Werlemark G, Matyasek R, Bringloe JB, Sieber V, El Mokadem H et al. (2005). Evolutionary implications of permanent odd polyploidy in the stable sexual, pentaploid of Rosa canina L. Heredity 94: 501–506.
Matyasek R, Tate JA, Lim YK, Srubarova H, Koh J, Leitch AR et al. (2007). Concerted evolution of rDNA in recently formed Tragopogon allotetraploids is typically associated with an inverse correlation between gene copy number and expression. Genetics 176: 2509–2519.
Nieto Feliner G, Rossello JA (2007). Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol Phylogenet Evol 44: 911–919.
Nybom H, Esselink GD, Werlemark G, Leus L, Vosman B (2006). Unique genomic configuration revealed by microsatellite DNA in polyploid dogroses, Rosa sect. Caninae. J Evol Biol 19: 635–648.
Nybom H, Esselink GD, Werlemark G, Vosman B (2004). Microsatellite DNA marker inheritance indicates preferential pairing between two highly homologous genomes in polyploid and hemisexual dogroses, Rosa L. Sect. Caninae DC. Heredity 92: 139–150.
Ritz CM, Schmuths H, Wissemann V (2005). Evolution by reticulation: European dogroses originated by multiple hybridization across the genus Rosa. J Heredity 96: 4–14.
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984). Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81: 8014–8018.
Täckholm G (1920). On the cytology of the genus Rosa. Svensk Bot Tidskrift 14: 300–311.
Volkov RA, Komarova NY, Hemleben V (2007). Ribosomal DNA in plant hybrids: inheritance, rearrangement, expression. Syst Biodivers 5: 261–276.
Weiss-Schneeweiss H, Schneeweiss GM, Stuessy TF, Mabuchi T, Park J-M, Jang C-G et al. (2007). Chromosomal stasis in diploids contrasts with restructuring in auto- and allopolyploid taxa of Hepatica (Ranunculaceae). New Phytol 174: 669–682.
Werlemark G, Nybom H (2001). Skewed distribution of morphological character scores and molecular markers in three interspecific crosses in Rosa sect. Caninae. Hereditas 134: 1–13.
Wissemann V (2002). Molecular evidence for allopolyploid origin of the Rosa canina-complex (Rosaceae, Rosoideae). J Appl Bot 76: 176–178.
Wissemann V (2003). Conventional taxonomy (wild roses). In Roberts AV, Debener T, Gudin S (eds). Encyclopedia of Rose Science. Elsevier Academic Press: Oxford, pp 111–117.
Wissemann V (2006). Beauty and the bastards. B I F Futura 21: 158–163.
Zhang LB, Comes HP, Kadereit JW (2001). Phylogeny and quaternary history of the European montane/alpine endemic Soldanella (Primulaceae) based on ITS and AFLP variation. Am J Bot 88: 2331–2345.
Zhang W, Qu J, Gu H, Gao W, Liu M, Chen J et al. (2002). Studies on the origin and evolution of tetraploid wheats based on the internal transcribed spacer (ITS) sequences of nuclear ribosomal DNA. Theor Appl Genet 104: 1099–1106.
Acknowledgements
The work was supported by the Academy of Sciences (AVOZ50040507 and AV0Z50040702), NERC (UK), Grant Agency of the Czech Republic (521/07/0116), MSMT (LC06004). We thank Ms Jana Kaiserlichova (CZ) for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Kovarik, A., Werlemark, G., Leitch, A. et al. The asymmetric meiosis in pentaploid dogroses (Rosa sect. Caninae) is associated with a skewed distribution of rRNA gene families in the gametes. Heredity 101, 359–367 (2008). https://doi.org/10.1038/hdy.2008.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2008.63
Keywords
This article is cited by
-
Do pentaploid hybrids mediate gene flow between tetraploid Senecio disjunctus and hexaploid S. carniolicus s. str. (S. carniolicus aggregate, Asteraceae)?
Alpine Botany (2021)
-
Hypo-hydroxymethylation of rRNA genes in the precocious Eriocheir sinensis testes revealed using hMeDIP-seq
Scientific Reports (2017)
-
Astonishing 35S rDNA diversity in the gymnosperm species Cycas revoluta Thunb
Chromosoma (2016)
-
DNA fingerprinting in botany: past, present, future
Investigative Genetics (2014)
-
Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids
BMC Genomics (2012)
