
Abstract
Mutations of the human desmin (DES) gene cause autosomal dominant and recessive myopathies affecting skeletal and cardiac muscle tissue. Desmin knockout mice (DES-KO), which develop progressive myopathy and cardiomyopathy, mirror rare human recessive desminopathies in which mutations on both DES alleles lead to a complete ablation of desmin protein expression. Here, we investigated whether an adeno-associated virus-mediated gene transfer of wild-type desmin cDNA (AAV-DES) attenuates cardiomyopathy in these mice. Our approach leads to a partial reconstitution of desmin protein expression and the de novo formation of the extrasarcomeric desmin–syncoilin network in cardiomyocytes of treated animals. This finding was accompanied by reduced fibrosis and heart weights and improved systolic left-ventricular function when compared with control vector-treated DES-KO mice. Since the re-expression of desmin protein in cardiomyocytes of DES-KO mice restores the extrasarcomeric desmin–syncoilin cytoskeleton, attenuates the degree of cardiac hypertrophy and fibrosis, and improves contractile function, AAV-mediated desmin gene transfer may be a novel and promising therapeutic approach for patients with cardiomyopathy due to the complete lack of desmin protein expression.
Similar content being viewed by others


Introduction
Desmin is a type III intermediate filament (IF) protein, which is abundantly expressed in smooth and striated muscle cells. In the latter, desmin—along with other IF proteins like syncoilin and synemin and the cytolinker protein plectin—forms a three-dimensional filamentous network, which interlinks neighboring myofibrils and connects the myofibrillar apparatus with the subsarcolemmal cytoskeleton at the level of intercalated discs, costameres, neuromuscular and myotendenious junctions as well as myonuclei and other cell organelles.1 The essential role of the extrasarcomeric desmin cytoskeleton in striated muscle tissue is highlighted by the observation that mutations of the human desmin (DES) gene on chromosome 2q35 cause hereditary and sporadic myopathies and cardiomyopathies.2, 3 The clinical spectrum of human desminopathies is highly variable. The disease onset ranges from the first to the eighth decade of life and the striated muscle pathology may manifest with pure myopathy, cardiomyopathy or a combination of both.2 Desmin-related cardiac disease manifestations comprise different forms of cardiomyopathy as well as various forms of often life-threatening cardiac conduction defects and arrhythmias.2 To date, no specific treatment exists for human desminopathies.
Since the first description of human DES mutations in 1998, over 70 myopathy and/or cardiomyopathy causing mutations have been reported.1 The vast majority of human desminopathies follow an autosomal dominant trait of inheritance and autosomal recessive cases have been published rarely so far.4 Out of the latter, few reports describe patients with myopathies and cardiomyopathies on the basis of homozygous or compound heterozygous DES mutations, which lead to a complete ablation of desmin protein expression.5, 6 In contrast to autosomal dominant desminopathies, these rare recessive desmin knockout patients have a much more unfavorable prognosis due to the development of cardiomyopathy, which usually leads to death in the second decade of life. Desmin-deficient (DES-KO) mice, which were described by two independent groups, recapitulate central disease aspects of desmin-deficiency in patients. DES-KO mice are viable and fertile, but develop clinical and morphological signs of a progressive myopathy and cardiomyopathy.7, 8, 9, 10
In the present study we addressed the issue if—and to what extent—an adeno-associated virus (AAV)-mediated gene transfer of wild-type (WT) DES-cDNA can prevent or attenuate the development of cardiomyopathy in DES-KO mice. Cardiac-specific AAV-mediated gene therapy approaches represent promising strategies to treat acquired and inherited cardiomyopathies. In dystrophin-deficient (mdx) and Sgcd-deficient (Sgcd-null) mice, animal models for human dystrophinopathy and sarcoglycanopathy, respectively, AAV-based gene replacement studies have even provided positive results as curative approaches.11, 12 With regard to the cardiac pathology in DES-KO mice, we used the AAV serotype 9 (AAV9), which exhibits the highest cardiac tropism in mice resulting in a homogeneous long-term transfer of intact cDNA throughout the heart.13, 14 We demonstrate that AAV9-mediated gene transfer of WT DES-cDNA leads to a partial reconstitution of desmin protein expression and marked improvement of morphological and functional parameters in hearts of DES-KO mice.
Results
Reconstitution of desmin expression and de novo formation of the extrasarcomeric desmin–syncoilin cytoskeleton in hearts of DES-KO mice
scAAV9-hTNNT2-mDES (n=10; AAV-Des) and scAAV9-CMV-MLC-hRluc (n=10; AAV-LUC) treated DES-KO mice were killed 10 months after the initial vector application. To assess the efficiency and molecular effects of our AAV-DES-mediated gene replacement approach, explanted hearts from treated DES-KO and untreated WT mice were analyzed by quantitative reverse transcriptase PCR, western blot and indirect immunofluorescence microscopy. Vector-mediated desmin expression in DES-KO mice attained 4.0±0.6% of the median WT mRNA levels and 22.2±9.8% of the median WT protein levels, respectively (Figures 1a–c).

Long-term desmin expression after intravenous injection of AAV-DES in DES-KO mice (n=10). (a) AAV-DES treatment resulted in desmin mRNA expression at levels of about 4.0±0.6% of those observed in wild-type mice (n=12). (b) Quantification of desmin protein expression revealed 22.2±9.8% of those in wild-type mice. (c) Detection of transgenic desmin expression in desmin-deficient mice (−/−, AAV-DES) in a representative western blot analysis (antibody DE-U-10). (d, e) Representative immunofluorescence staining of plasma membranes (green), nuclei (blue) and desmin (red, antibody DE-U-10) of an AAV-LUC-treated desmin-deficient mouse (n=10) (d), an AAV-DES-treated desmin-deficient mouse (n=10) (e) and a wild-type control (n=12) (f). Note that individual cardiomyocytes in AAV-DES-treated mice display variable levels of transgenic desmin protein expression with a cross-striated staining pattern and a signal accumulation at the level of intercalated discs.
The re-expression of desmin protein was further mirrored in our indirect immunofluorescence analysis, which showed a re-formation of the characteristic cross-striated desmin cytoskeletal network in cardiomyocytes of treated DES-KO mice (Figures 1d–f).
Furthermore, syncoilin and plectin stainings demonstrated the functionality of vector-mediated desmin. Previous studies in striated muscle of DES-KO mice showed that the ablation of desmin alters the subcellular distribution of syncoilin but not of plectin.15, 16 In this context, we observed that the vector-mediated reconstitution of the desmin filamentous network increased the level of syncoilin protein expression and restored its typical staining pattern in cardiomyocytes (Figures 2).

Desmin/plectin/syncoilin co-staining with 4',6-Diamidin-2-phenylindol (DAPI). First column: (a, d) wild type (n=3). Second column: (b, e) AAV-LUC-treated desmin-deficient mouse (n=4). Third column: (c, f) and detail of (f) (x) syncoilin, y desmin, z merge) AAV-DES-treated desmin-deficient mouse (n=4). First row (a–c): desmin green, plectin red. Second row and details of (f) (d–f, x–z): desmin red, syncoilin green. While the subcellular distribution of plectin seems unaffected by the absence of desmin, the re-expression of desmin has a dramatic effect on the syncoilin staining pattern. Note that the AAV-DES treatment leads to the de novo formation of a cross-striated desmin–syncoilin staining pattern in a subset of DES-KO cardiomyocytes (x–z).
Reduced ventricular fibrosis and hypertrophy
Untreated, DES-KO mice phenotypically show cardiomyopathy with increased cardiac fibrosis and enlarged hearts compared with WT littermates. Heart weight normalized to tibia length is an established marker for cardiac hypertrophy or dilation in dystrophic mouse models.17 Heart weight to tibia length ratios were significantly (P<0.03) reduced in AAV-DES-treated DES-KO mice, indicating a decreased normalized heart weight similar to WT controls (AAV-DES-treated DES-KO mice: 8.9±1.1 mg mm−1; WT mice: 9.2±1.1 mg mm−1; AAV-LUC-treated DES-KO mice: 11.3±1.0 mg mm−1; Figures 3a and b). Cardiomyocyte cross-sectional areas were also decreased in AAV-DES-treated animals compared with the AAV-LUC group, measuring 219±14 and 272±27 μm2 (P=0.1), respectively, pointing towards a decrease in hypertrophy in AAV-DES-treated animals.

Morphometric and histological analysis of hearts from mice treated with AAV-DES compared with AAV-LUC and wild-type controls. (a) Heart weight to tibia length ratios are significantly decreased in AAV-DES-treated animals (n=10) when compared with AAV-LUC controls (n=9) or wild-type mice (n=12). (b) Ventricular fibrosis is also significantly decreased in AAV-DES (n=10) and wild type (n=12) when compared with AAV-LUC (n=10). (c–e) Representative Masson’s trichrome stainings of the left ventricle of AAV-LUC (n=10) (c), AAV-DES (n=10) (d) and wild type (n=12) (e). Note the intense fibrotic area (blue) depicted in (c). *P<0.05, **P<0.01.
In order to evaluate the cardioprotective effect of our treatment, ventricular tissue was further analyzed for fibrosis. Masson’s trichrome stained sections revealed large fibrotic areas in AAV-LUC animals (Figure 3c), which were not present in AAV-DES animals or WT controls (Figures 3d and e). Quantification using standardized automated image analysis yielded a significant reduction in ventricular fibrous tissue in AAV-DES animals (P<0.005) with 14.3±3.2% in AAV-LUC mice, 5.0±0.7% in AAV-DES animals and 5.1±1.2% in WT.
Improved left-ventricular systolic function and reduction of BNP levels
The cardioprotective effects of the chosen AAV-DES gene therapy approach were assessed in vivo using serial echocardiography and, finally, measurements of PVL.
AAV-LUC-treated animals developed progressive dilated cardiomyopathy with impaired left-ventricular systolic function. In comparison with WT animals they showed a progressive decrease in fractional shortening (FS) and a substantial increase in left-ventricular end-diastolic diameter. A significant decrease in FS of AAV-LUC mice was first noted at 6 months after vector injection. In contrast, AAV-DES-treated animals exhibited an attenuated decrease in FS and practically no increase in left-ventricular end-diastolic diameter at the end of this 10 month study (Figure 4 and Supplementary Table S1). With an FS of 46.4±4.3%, left-ventricular function was significantly better in AAV-DES mice than in AAV-LUC control-treated animals (32.2±6.1%), but did not reach WT levels (62.5±1.6%). Representative echocardiographic findings of AAV-LUC, AAV-DES and WT mice are also depicted in Supplementary Video S1.

Follow-up echocardiography. Mice were injected at the age of 2 months following baseline echocardiography. (a) Systolic left-ventricular (LV) function was significantly reduced in the AAV-LUC group (n=9) after 12 months in comparison with AAV-DES-treated knockout mice (n=10). (b) LV end-diastolic diameter in the AAV-LUC group was significantly increased when compared with WT (n=12) controls, while no significant differences were detectable between the AAV-DES group and WT controls (P=0.7). *P<0.05, ***P<0.001. † premature death of an AAV-LUC animal.
The improvement of functional parameters in AAV-DES-treated animals was further mirrored by our PVL experiments. Here, end-systolic pressure was significantly decreased in AAV-LUC-treated animals compared with WT mice (84.8±6.7 vs 103.3±2.0 mmHg, P<0.04) with a significant improvement in AAV-DES-treated mice (109.3±5.3 mmHg, P<0.02). Stroke work, a further parameter for systolic capacity, was also significantly increased in AAV-DES-treated mice when compared with AAV-LUC-treated mice (P<0.02; Supplementary Table S2). Moreover, AAV-DES-treated mice exhibited a significantly higher maximal rate of pressure development (dp/dtmax) than AAV-LUC (Figure 5).

Pressure volume loop measurements. AAV-LUC controls (n=9) showed a significantly impaired systolic left-ventricular function, noticeably reduced end-systolic pressure (ESP, a), decreased stroke work (SW, b) and lower pressure development rates (dP/dtmax, c) when compared with wild-type mice (n=7) and AAV-DES-treated animals (n=8). *P<0.05.
Invasive analysis with PVL further revealed a significant impairment of passive and active relaxation of the left ventricle in AAV-LUC-treated mice compared with WT controls (dp/dtmin and tau Glantz, P<0.001 and P<0.01, respectively). Although there was a trend towards increased dp/dtmin and accelerated tau Glantz values in the AAV-DES group compared with control-treated DES-KO mice, differences did not reach statistical significance.
In addition to hemodynamic measurements, left-ventricular BNP mRNA expression levels as a marker for heart failure were measured. Both AAV-LUC- and AAV-DES-treated mice exhibited significantly increased left-ventricular BNP mRNA expression compared with WT. However, BNP expression was markedly reduced in AAV-DES-treated mice compared with AAV-LUC controls (5.7±0.7 vs 8.1±0.9 fold WT expression).
Discussion
Autosomal recessive cardiomyopathies and myopathies based on a complete lack of desmin protein expression are a rare subgroup of human desminopathies with a poor prognosis due to progressive cardiac failure. Since no causative therapy is currently available for this disorder, we investigated if and to what extent an AAV9-mediated gene transfer of WT desmin cDNA may ameliorate the cardiac pathology in DES-KO mice. Using a dose of 3 × 1012 vector genomes, this approach led to a reconstitution of desmin expression with 4.0±0.6 and 22.2±9.8% of WT mRNA and protein levels, respectively. The discrepancy between vector-mediated protein and mRNA desmin expression is impressive and probably due to the SV40 poly(A) domain added in the 3′UTR, which is known to further stabilize mRNA and enhance translation.18, 19
On the level of individual cardiomyocytes, this treatment resulted in the de novo formation of the three-dimensional desmin network with a staining pattern indistinguishable from that observed in WT animals. Importantly, the re-formation of the desmin network in hearts of DES-KO mice also resulted in proper subcellular re-localization of the IF protein syncoilin. Together, these findings indicate that—even at a rather low desmin protein expression level—the chosen strategy can indeed restore the structural formation of the pivotal extrasarcomeric desmin–syncoilin cytoskeleton in cardiomyocytes.
The beneficial long-term treatment effects of the AAV-DES application were also mirrored on the morphological and functional level. With interindividual variation in onset and intensity, DES-KO mice develop fibrosis, hypertrophy, compromised systolic function and eventually cardiac dilation.9, 20 Since the phenotype in control-treated mice corresponded to these previous reports in DES-KO mice and we could not observe any effect of neither the promoter nor reporter gene on cardiac function before, we do not expect any effect of the reporter gene or viral vector itself.21 Although we have used a CMV-MLC-promoter for the control AAV vector and a more specific human troponin T promoter for cardiac-specific expression of the desmin cDNA, there is no major limitation since the only difference between the two promoters is a mild hepatic expression of the luciferase reporter.21
In contrast to AAV-LUC control-treated animals, AAV-DES-treated mice displayed reduced ventricular fibrous tissue, ventricular dilation, hypertrophy, decreased BNP expression and an improvement of left-ventricular systolic function parameters despite only 22.2±9.8% desmin protein levels compared with WT mice. Similarly, a threshold of 4% of dystrophin protein was identified as sufficient to ameliorate cardiac function in transgenic mice with low levels of dystrophin expression (3–15%).22 Nevertheless, since mice were not challenged in our study, it remains to be elucidated whether the levels of overexpression would be sufficient to compensate also for physical stress.
However, the achieved desmin re-expression did not completely rescue, but rather ameliorated cardiac involvement in DES-KO mice, as WT littermates still exhibited overall superior cardiac contractile parameters. The partial therapeutic effect might be explained by either irreversible preformed ultra-structural damage prior to therapy or an extent of desmin re-expression still too low for a complete rescue of the phenotype.20 With regard to the latter, a further increase in the AAV-DES vector dose or further improvements of the vector cassette might increase the transgenic desmin expression.23, 24, 25 Since the human troponin T promoter used in this study previously resulted in successful transduction of the vast majority of cardiomyocytes after systemic gene transfer with AAV vectors into adult mice,21 inclusion of further untranslated 5′ or 3′ regions of the desmin cDNA might be helpful to increase the efficiency of protein expression. Furthermore, injection in even younger mice as previously shown for a similar AAV9/troponin T promoter approach in a murine model of hypertrophic cardiomyopathy might not only enable a higher transduction efficiency by using higher titers per body weight, but could also prevent progressive cardiac damage during adolescence.26
The observed partial rescue combined with variations in disease onset and severity in DES-KO mice might also explain the long time interval between transgene desmin expression, which was detected 4 weeks after injection, and hemodynamically significant differences between AAV-DES and AAV-LUC treated animals. DES-KO mice living a sedentary life style show much variation in onset and severity of dilated cardiomyopathy evident in the large variance in echocardiographic findings specifically in the AAV-LUC group as well as in the premature death of a AAV-LUC animal suffering from dilated cardiomyopathy at a time point when others still seemed healthy. Future studies are necessary to elucidate whether streneous physical exercise by forced wheel running or swimming might aggravate cardiomyopathy and thus reduce variations.9, 27
Taken together, our study demonstrates that an AAV9-mediated gene transfer of WT desmin cDNA results in the re-expression of desmin protein, the de novo formation of the essential extrasarcomeric desmin–syncoilin IFs system in cardiomyocytes as well as an improvement of the structural and functional pathology of hearts of DES-KO mice. Thus, AAV9-mediated desmin gene transfer may be a novel and promising therapeutic approach for patients with cardiomyopathy due to the complete lack of desmin protein expression.
Materials and methods
Animal procedures and study protocol
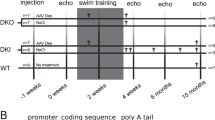
Two-month-old DES-KO (B6.129S2/Sv-Destm1Cba/Orl) male mice6 were randomly assigned to the treatment (AAV-DES, n=10) or vector control group (AAV-LUC, n=10). Randomization was performed allocating cages to the AAV-DES or AAV-LUC using a javascript random number generator. The first randomly calculated combination yielding equal group sizes was applied. A dose of 3 × 1012 vector genomes was systemically administered through the tail vein. Twelve genetically healthy littermates were used as WT controls. Genotyping was performed as described by Torrente et al. (Supplementary Methods).28 Animals were fed ad libitum and housed in a temperature- and humidity-controlled room in a specified pathogen-free environment under 12:12 h light/dark cycles. All procedures involving the use and care of animals were performed according to the Directive 2010/63/EU of the European Parliament and the German animal protection code. Approval was granted by the local ethics review board (G143/11). Left ventricular function was assessed using transthoracic echocardiography before vector application and every 3 months following treatment. Final analysis was performed with echocardiography and PVL 10 months after vector application, upon which mice were killed. Mice were killed by cervical dislocation and weighed. Hearts were removed, washed in phosphate-buffered saline (PBS), weighed and cut in half biventricularily along the long axis of the left ventricle. One half was embedded in tissue-tek (Sakura, Staufen, Germany) and frozen on dry ice. The other half was further dissected. Atria and right ventricle were removed and flash frozen. Additional samples from liver and soleus muscle were taken. The right hindlimb was amputated at the femur level and incubated in a 37% hydrogen peroxide solution for 2 weeks. Tibia lengths were measured with a slide gauge.
All measurements were obtained by a blinded investigator. One animal of the AAV-LUC control group suffering from severe dilated cardiomyopathy died in the course of the study. Preliminary survival analyses showed a similar trend with a sharp increase in mortality in desmin-deficient mice after week 56 (Supplementary Figure S1).
Cloning of the murine cDNA encoding desmin
RNA was isolated from the ventricles of a C57/B6 mouse (Charles River, Sulzfeld Germany) using the RNeasyFibrous Tissue Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. RNA was transcribed into cDNA with the Superscript III kit (Invitrogen, Darmstadt, Germany). Two desmin coding sequences were amplified using phusion polymerase (Finnzymes, Vantaa, Finland) applying a two-step protocol (98 °C; 72 °C; 30 cycles) with the same forward (GGATCCACCGGTGCCACCATGAGCCAGGCCTACTCGTCC), but different reverse primers (TGAAGCGTACAAGAGGTGGCTGAGGGGTTCCCTGG and TGAAGCTGTACAGTGAGGACGGGGCCAGGACACTGAA) creating one clone with and one without an extended 3′UTR. Amplificons were subsequently cloned into the pCR blunt vector (zero blunt cloning kit; Invitrogen) creating pCR-Blunt-mDES-ext and pCR-Blunt-mDES, respectively. Plasmids from resulting colonies were harvested (QIAprep Spin Miniprep Kit; Qiagen) and sequenced (MWG, Ebersberg, Germany).
Generation of AAV vectors
Murine desmin cDNA was inserted with BamHI/BsrGI into a self-complementary AAV vector genome plasmid, derived from pdsCMV-MLC0.26-EGFP,29 under control of the human troponin T promoter (hTNNT2), resulting in pds_hTNNT2-mDes. AAV9 vectors for expression of desmin or luciferase (control) were generated by cotransfection of pds_hTNNT2-mDES or pdsCMV-MLC-hRluc together with pDP9rs, a derivate from pDP2rs30 with the AAV9 cap gene from p5E18-VD2–9,31 in Hek293T cells using polyethylenimine (Sigma Aldrich, Taufkirchen, Germany). AAV was purified and titrated as previously described.21
Western blot
Tissue was homogenized in radioimmunoprecipitation assay buffer and protein concentrations were measured using the DC Protein Assay (BioRad, Munich, Germany) following the manufacturer’s instructions for microtiter plates. Samples containing 100 μg of protein were diluted in protein sample buffer and heated for 5 min at 95 °C. Electrophoresis was carried out using a 4–20% sodium dodecyl sulfate glycine gradient gel (Invitrogen or BioRad) at 120 V for 2 h. Gels were blotted to PVDF (immobilon, Merck Millipore, Darmstadt, Germany) or nitrocellulose (Whatman Protran BA83; Sigma Aldrich) membranes in modified Towbin buffer applying 300 mA at 4 °C overnight. Membranes were blocked with milk powder (5% solved in TBS-T) for 1 h. Primary antibodies for desmin (DE-U-10; Sigma Aldrich, 1:200), GAPDH (Sigma Aldrich, 1:7500), plectin (antiserum #9; G. Wiche, MFPL, University of Vienna, Vienna, Austria, 1:3000) and syncoilin (sc-162284; Santa Cruz Biotechnology, Dallas, TX, USA, 1:500) were solved in blocking buffer and membranes were incubated for 1 h. Secondary antibodies goat-anti-mouse-HRP (Santa Cruz Biotechnology), goat-anti-rabbit-HRP (Santa Cruz Biotechnology) and rabbit-anti-goat-HRP (BioRad) were applied for 45 min at room temperature at a dilution of 1:5000. Membranes were washed thoroughly in TBS-T after each antibody staining as well as after antibody stripping (Thermo Scientific, Rockford, IL, USA) performed between desmin and GAPDH staining. Blots were read out using the ChemiDoc imaging system (BioRad) applying Pierce ECL solution (Thermo Scientific) following the manufacturer’s instructions. Data acquisition and quantification were performed with ImageLab and ImageJ. Expression levels were normalized to GAPDH and quantified relative to a WT standard.
Quantitative reverse transcriptase PCR
RNA was isolated from samples using the RNeasyFibrous Tissue Mini Kit (Qiagen), optional DNase digest was performed, 0.6 μg RNA was transcribed into cDNA with the help of the Superscript III Kit (Invitrogen) and RNA digestion was performed using RNase H. Commonly used primers were employed for GAPDH and BNP quantification, while a primer blast search was performed in order to find suitable primers for desmin (NM_010043.1) quantification (Supplementary Table S3). Desmin primers were designed to distinguish between WT and vector-mediated desmin expression making use of different 3′ UTR sequences. cDNA was quantified using iTaq Universal SYBR Green Supermix (BioRad) on a CFX96 Touch Real-Time PCR detection system (BioRad) applying a common two-step protocol (60 °C; 95 °C; 30 cycles). Two technical replicates were run for each reaction. Expression levels of WT and vector-mediated desmin were quantified using plasmid standards: pCR-Blunt-mDes-ext served as WT standard and dsAAV-hTNNT2-mDES as a transgene standard. BNP expression was quantified using the ΔΔct method normalizing to GAPDH.
Immunofluorescence
Nine micrometer cryosections were fixed in 3% PFA dissolved in 0.1 m phosphate buffer for 20 min, washed thrice in PBS and plasma membranes were stained with wheat germ agglutinin (Molecular Probes, Invitrogen) at a concentration of 5 μg ml−1 for 10 min followed by washing in PBS. Cells were permeablized in 0.2% Triton X-100 (Invitrogen) in PBS for 5 min at room temperature and washed three times. Alternatively, 5 μm cryosections were fixed with ice-cold acetone for 10 min and air-dried prior to blocking. Slides were blocked in PBS containing 10% fetal calf serum and 1% goat serum for 1 h and incubated with primary antibodies for desmin (DE-U-10, diluted 1:50, for the co-staining with wheat germ agglutinin), desmin (D33; Dako, Hamburg, Germany, 1:100), plectin (GP-21; Progen, Heidelberg, Germany, 1:400), and syncoilin (sc-162284; Santa Cruz Biotechnology, 1:100) in PBS containing 10% fetal calf serum overnight at 4 °C in a wet chamber. Samples were washed, incubated with donkey-anti-mouse-IgG (DaM IgG AF647; Jackson ImmunoResearch Laboratories, Baltimore, PA, USA, diluted 1:100), goat-anti-guinea pig-IgG (Alexa Fluor 555; Life Technologies, 1:200), goat-anti-mouse IgG (Cy2; Jackson ImmunoResearch Laboratories, 1:200), donkey-anti-mouse-IgG (Rhodamine red; Jackson ImmunoResearch Laboratories, 1:200), or donkey-anti-goat (Dylight 649; Thermo Scientific,1:200) antibodies in PBS containing 10% fetal calf serum for 1 h. Slides were washed thoroughly, mounted (Vectashield mounting medium with 4',6-Diamidin-2-phenylindol (DAPI); Vector, Burlingame, UK) and analyzed by fluorescence microscopy (Ni-E for wide field imaging and A1R for confocal imaging; Nikon, Düsseldorf, Germany) using a DS-Qi1Mc camera (Nikon) and a 32-channel spectral detector (Nikon), respectively. Alternatively, a confocal laser scanning microscope (Zeiss, Oberkochen, Germany) equipped with a Plan-Apochromat 63 × 1.4NA objective lens was used. Images from wheat germ agglutinin stained slides were acquired on an Olympus IX81 microscope.
Masson’s trichrome staining
Slides were thawed, fixed in 10% formalin for 10 min at room temperature, washed, dipped four times in Harris’ hematoxylin (Sigma Aldrich) and incubated for 3 min in Mallory red (0.5% fuchsin acid, 0.5% glacial acetic acid). After brief washing in deionized water, slides were put in phosphotungstic acid (2.5%) for 15 min and stained in methyl blue (0.5% methyl blue, 2.5% glacial acetic acid) for 45 s. Dehydration was performed with graded alcohols in ascending order. Slides were subsequently placed in acidified ethanol containing 1% glacial acetic acid for 5 min. Slides were put in xylene for 5 min, mounted (Roti histo kit II, Roth), and analyzed on a Ni-E microscope (Nikon) using a DS-Ri1 camera (Nikon).
Image analysis
Fibrosis was measured using stitched images taken at × 4 magnification on the Ni-E microscope applying a macro. Images were color-thresholded in ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) using the HSB filter sets dividing the image into two-color ranges (Hue) from 0 to 199 and 200 to 255 and excluding less saturated (S) pixels (0–35). Both image masks were each overlaid on the original image as a manual quality control. Particles on the created masks were subsequently analyzed and the total areas were used to create the blue to total ratio (blue/(blue+red) × 100) indicating the percentage of fibrous tissue.
Transthoracic echocardiography
Echocardiographies were performed with hand restrained mice using the Sonos 5500 ultrasound imaging system (Agilent, Santa Clara, CA, USA) with the 15-6L (Agilent) probe and individually adjusted optimal device settings. M-Mode short axis (SAX) cine loops were obtained. The left-ventricular end-diastolic and end-systolic diameters (LVEDD, LVESD) of three consecutive heartbeats were determined. Means were established and the FS was calculated.
Pressure volume loops
Animals were anesthetized using 0.4 mg kg−1 medetomidine, 5 mg kg−1 midazolam and 500 μg kg−1 fentanyl injected intraperitoneally. Body temperature was maintained at 37 °C using a homeothermic blanket (Harvard Apparatus, Edenbridge Kent, UK). Mice were catheterized through the left carotid artery with a 1.2 Fr catheter and PVL were measured in the left ventricle. Left ventricular volumes were extrapolated from admittance magnitude and admittance phase in real time using the ADVantage PV system (Scisense, Transonic, Maastricht, The Netherlands). Pressure and volume data were recorded using a Scisense 404-16 Bit Four Channel Recorder (Scisense) with LabScribe2 Software (Scisense). Indices of systolic function included end-systolic pressure, stroke volume and stroke work. Load-dependent myocardial contractility was assessed by maximal rate of pressure development (dP/dtmax). Transient inferior vena caval compressions were applied to reduce preload and determine end-systolic elastance as a parameter of load-independent myocardial contractility as previously described by Bauer et al.32 Diastolic function was assessed by end-diastolic pressure, tau (Glantz method, regression of log of pressure vs time) and the maximal rate of pressure decay (dP/dtmin).
Statistics
All data are expressed as mean±standard error. Statistical analyses were performed using R.33 Unless indicated otherwise, pairwise two-sided t-tests were used for statistical testing applying the Holm–Bonferroni algorithm for P-value adjustment. P-values under 0.05 were considered significant. The Shapiro–Wilk test was used to test for normal distribution and the and the F-test was used to test for similar variances.
References
Clemen CS, Herrmann H, Strelkov SV, Schröder R . Desminopathies: pathology and mechanisms. Acta Neuropathol 2013; 125: 47–75.
van Spaendonck-Zwarts K, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ et al. Desmin-related myopathy: a review and meta-analysis. Clin Genet 2011; 80: 354–366.
Viegas-Péquignot E, Li ZL, Dutrillaux B, Apiou F, Paulin D . Assignment of human desmin gene to band 2q35 by nonradioactive in situ hybridization. Hum Genet 1989; 83: 33–36.
Goldfarb LG, Dalakas MC . Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009; 119: 1806–1813.
Carmingac V, Sharma S, Arbogast S, Fischer D, Serreri C, Serria M et al. A homozygous desmin deletion causes an Emery-Dreifuss like recessive myopathy with desmin depletion. Neuromuscul Discord 2009; 19: 600.
Henderson M, De Waele L, Hudson J, Eagle M, Sewry C, Marsh J et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol 2013; 125: 917–919.
Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y . Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 1996; 134: 1255–1270.
Schrickel JW, Stöckigt F, Krzyzak W, Paulin D, Li Z, Lübkemeier I et al. Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J Interv Card Electrophysiol 2010; 28: 71–80.
Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C et al. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol 1999; 31: 2063–2076.
Li Z, Colucci-Guyon E, Pinçon-Raymond M, Mericskay M, Pournin S, Paulin D et al. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol 1996; 175: 362–366.
Schinkel S, Bauer R, Bekeredjian R, Stucka R, Rutschow D, Lochmüller H et al. Long-term preservation of cardiac structure and function after adeno-associated virus serotype 9-mediated microdystrophin gene transfer in mdx mice. Hum Gene Ther 2012; 23: 566–575.
Goehringer C, Rutschow D, Bauer R, Schinkel S, Weichenhan D, Bekeredjian R et al. Prevention of cardiomyopathy in delta-sarcoglycan knockout mice after systemic transfer of targeted adeno-associated viral vectors. Cardiovasc Res 2009; 82: 404–410.
Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 2006; 14: 45–53.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE . Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 2008; 16: 1073–1080.
Carlsson L, Li ZL, Paulin D, Price MG, Breckler J, Robson RM et al. Differences in the distribution of synemin, paranemin, and plectin in skeletal muscles of wild-type and desmin knock-out mice. Histochem Cell Biol 2000; 114: 39–47.
McCullagh KJA, Edwards B, Poon E, Lovering RM, Paulin D, Davies KE . Intermediate filament-like protein syncoilin in normal and myopathic striated muscle. Neuromusc Disord 2007; 17: 970–979.
Yin FC, Spurgeon HA, Rakusan K, Weisfeldt ML, Lakatta EG . Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am J Physiol 1982; 243: H941–H947.
Wang H, Sun W, Li Z, Wang X, Lv Z . Identification and characterization of two critical sequences in SV40PolyA that activate the green fluorescent protein reporter gene. Genet Mol Biol 2011; 34: 396–405.
Xu ZL, Mizuguchi H, Ishii-Watabe A, Uchida E, Mayumi T, Hayakawa T . Optimization of transcriptional regulatory elements for constructing plasmid vectors. Gene 2001; 272: 149–156.
Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D . Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol 1997; 29: 2107–2124.
Werfel S, Jungmann A, Lehmann L, Ksienzyk J, Bekeredjian R, Kaya Z et al. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV-mediated expression of Cre recombinase. Cardiovasc Res 2014; 104: 15–23.
van Putten M, van der Pijl EM, Hulsker M, Verhaart IEC, Nadarajah VD, van der Weerd L et al. Low dystrophin levels in heart can delay heart failure in mdx mice. J Mol Cell Cardiol 2014; 69: 17–23.
Li ZL, Paulin D . High level desmin expression depends on a muscle-specific enhancer. J Biol Chem 1991; 266: 6562–6570.
Price MG . Molecular analysis of intermediate filament cytoskeleton-a putative load-bearing structure. Am J Physiol 1984; 246 (Pt 2): 566–572.
Talbot GE, Waddington SN, Bales O, Tchen RC, Antoniou MN . Desmin-regulated lentiviral vectors for skeletal muscle gene transfer. Mol Ther 2010; 18: 601–608.
Mearini G, Stimpel D, Geertz B, Weinberger F, Kramer E, Schlossarek S et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun 2014; 5: 5515.
Costas JM, Nye DJ, Henley JB, Plochocki JH . Voluntary exercise induces structural remodeling in the hearts of dystrophin-deficient mice. Muscle Nerve 2010; 42: 881–885.
Torrente Y, Tremblay JP, Pisati F, Belicchi M, Rossi B, Sironi M et al. Intraarterial injection of muscle-derived CD34(+)Sca-1(+) stem cells restores dystrophin in mdx mice. J Cell Biol 2001; 152: 335–348.
Müller OJ, Schinkel S, Kleinschmidt JA, Katus HA, Bekeredjian R . Augmentation of AAV-mediated cardiac gene transfer after systemic administration in adult rats. Gene Therapy 2008; 15: 1558–1565.
Grimm D, Kay MA, Kleinschmidt JA . Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther 2003; 7: 839–850.
Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol 2004; 78: 6381–6388.
Bauer R, Blain A, Greally E, Bushby K, Lochmüller H, Laval S et al. Intolerance to ss-blockade in a mouse model of delta-sarcoglycan-deficient muscular dystrophy cardiomyopathy. Eur J Heart Fail 2010; 12: 1163–1170.
Core Team. R . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2012.
Acknowledgements
We thank Ulrike Gärtner from the animal core unit of the University of Heidelberg for expert support in animal experiments. We further would like to thank Denise Paulin (Institut de Biologie Paris Seine, Université Pierre et Marie Curie) for critically reading the manuscript. This work was supported by the German Research Foundation (DFG) within the framework of the multi-location research group FOR1228 (grants CL 381/7-1 to CSC, MU 1654/8-1 to OJM and SCHR 562/13-1 to RS).
Author contributions
OJM, RB and MBH jointly conceived the study, reviewed all data, prepared the figures and wrote the manuscript. MBH, AJ, LW, K-HS and CSC conceived and carried out experiments and analyzed data. ZL provided desmin knockout mice. RS, KR and HAK conceived experiments, interpreted data and wrote the manuscript. All authors had final approval of the submitted and published versions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Heckmann, M., Bauer, R., Jungmann, A. et al. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther 23, 673–679 (2016). https://doi.org/10.1038/gt.2016.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2016.40
This article is cited by
-
AAV-mediated expression of NFAT decoy oligonucleotides protects from cardiac hypertrophy and heart failure
Basic Research in Cardiology (2021)
-
Cardiac transcriptional and metabolic changes following thoracotomy
Scientific Reports (2020)
-
AAV-mediated cardiac gene transfer of wild-type desmin in mouse models for recessive desminopathies
Gene Therapy (2020)
-
Molecular insights into cardiomyopathies associated with desmin (DES) mutations
Biophysical Reviews (2018)

{kind=link}