

Key Points
- Gastroesophageal reflux disease (GERD) includes all consequences of reflux of acid or other irritants from the stomach into the esophagus. The main cause of gastroesophageal reflux is incompetence of the antireflux barriers at the esophagogastric junction.
- Gastric pepsin duodenal contents exacerbate the action of acid and deleterious effect on the production of esophagitis.
- The antireflux barriers include two "sphincter" mechanisms: the lower esophageal sphincter (LES), and the crural diaphragm that functions as an external sphincter.
- Gastroesophageal reflux occurs when LES pressure is lower than the intragastric pressure such as in LES hypotension, increased frequency of transient lower esophageal sphincter relaxation (TLESR), when the intragastric pressure increases.
- The severity of GERD increases progressively with reflux that is mainly in the postprandial period to that in the upright posture, to that in the supine or that is bipositional reflux. Nighttime reflux leads to severe GERD.
- Hiatal hernia results from multiple mechanisms and is associated with a decreased LES pressure, decreased acid clearance, increased reflux, and more severe esophagitis.
- Mucosal defense mechanisms may be overcome by prolonged exposure of the esophageal mucosa to a pH <4 that may lead to severe and complicated esophagitis.
- Esophageal mucosal inflammation may affect nerves and muscle that alter LES function and esophageal body motility. A vicious cycle of inflammation and impaired motility may cause progressive disease.
- Patients with GERD may develop endoscopically visible erosive esophagitis or endoscopically negative nonerosive or negative endoscopy reflux disease (NERD). In NERD, factors such as visceral hypersensitivity or more proximal reflux of acid or nonacid material may be important. Acid and inflammatory mediators may gain access to sensory pathways and produce symptoms either by a direct action on the nerves or by producing abnormal muscle contraction.
Introduction
The factors generally accepted as important for the development of gastroesophageal reflux disease (GERD) have been well documented (Table 1).1, 2, 3 Abnormality of any one of these factors holds the potential to disturb the normal equilibrium. These factors include gastric acid and other refluxed contents; delayed gastric emptying; structural and physiologic antireflux mechanisms at the gastroesophageal region; transient lower esophageal sphincter relaxation (TLESR); esophageal clearance mechanisms; ingested irritants; ingested substances that alter gastric, lower esophageal sphincter (LES), or esophageal motor function; mucosal integrity and defense mechanisms; visceral hypersensitivity; and genetic factors.

Despite the many factors that operate, four main fundamental factors stand out as most important: (1) gastric acid; (2) the structural integrity, function, and competence of the LES that either prevent or allow reflux; (3) the esophageal mucosal defense mechanisms that are primarily called into play when there is excess exposure of the mucosa to gastric acid; and (4) the sensory mechanisms that speak to symptoms. That is, an incompetent gastroesophageal reflux mechanism allows abnormal amounts of gastric acid to enter the esophagus, where the acid burden causes mucosal damage and/or symptoms (Figure 1). When mucosal damage occurs (esophagitis), a vicious cycle can ensue to accentuate and maintain the GERD (Figure 2). Then both more acid reflux and decreased clearance of the acid can prolong contact of the acid with the esophageal mucosa.
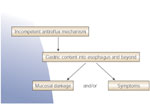
Figure 2: Gastroesophageal reflux disease initiates a vicious cycle of increasing esophageal acid exposure.
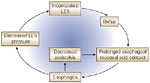
LES, lower esophageal sphincter. (Source: AGA teaching unit 7, Neurogastroenterology and Motility: Upper GI Tract, slide 34., used with permission. Copyright American Gastroenterological Association, Bethesda, MD.)
The GERD category also encompasses a group of patients that have nonerosive or negative endoscopy reflux disease (NERD). In these patients, esophageal acid exposure may be normal and factors such as visceral hypersensitivity or more proximal reflux of acid or nonacid material may be important.2, 4
Gastric Acid and Other Refluxed Substances
Gastric Acid
The normal physiology of acid secretion is well understood, and is not discussed here. Reflux of acid is the dominant irritant to the esophagus in the development and progression of GERD, although the presence of bile and other compounds in gastric juice may contribute to the "reflux burden" when combined with the acid or on their own.5 The reflux burden of acid requires the presence of acid secretion, and is further determined by dysfunction of the gastroesophageal competence mechanism that allows increased reflux, and by decreased esophageal clearance that increases contact time of acid with the mucosa.6 However, there are a number of relevant issues that arise in regard to gastric acid secretion and the development of GERD. Two issues have attracted considerable attention and controversy: (1) Is GERD associated with increased acid secretion? and (2) Does the effect of Helicobacter pylori infection on acid secretion impact on the development of GERD?
Levels of Gastric Acid Secretion
Acid suppression results in successful treatment of esophagitis.7 This response has raised the possibility that elevated levels of acid secretion would increase the "acid burden" that impacts on the esophagus and could be associated with the propensity to develop esophagitis, whereas decreased gastric acid secretion could result in a decrease in GERD and its complications. The arguments in this regard have surfaced recently in relation to the effects of H. pylori infection on acid secretion before and after the treatment of the infection, and the prevalence and development of esophagitis.6, 8, 9, 10, 11, 12, 13, 14, 15
Despite the intuitive logic that increased gastric acid secretion should increase the prevalence of esophagitis, there is little or no evidence to support this conclusion for the usual cases of GERD. Marked increases in gastric secretion as with Zollinger-Ellison syndrome are an exception. In an earlier study, the presence of esophagitis was not associated with elevated levels of acid secretion or with a lower gastric pH than in normal subjects.16 A more recent study indicated that mild erosive esophagitis can occur in the presence of decreased as well as normal acid secretion,17 and in the absence of Barrett's esophagus, acid secretion in patients with esophagitis is not different from that in normal subjects.18 This information suggests that beyond a certain level of gastric acid secretion, other factors are more important in determining the "acid burden" that produces esophagitis or other manifestations of GERD.
H. Pylori and Gastric Acid
On the other hand, there is evidence accumulating that decreased acid secretion with H. pylori infection of the stomach or atrophic gastritis is associated with decreased prevalence of esophagitis. This conclusion derives from many studies that demonstrate that the prevalence of H. pylori infection is less in patients with esophagitis (Figure 3).6, 12 For example in Japan, the prevalence of H. pylori is as high as 71% in asymptomatic subjects, but only 30% in patients with esophagitis, and is reported as low as 0% in patients with long segment Barrett's.18 A similar decreased prevalence of H. pylori in patients with esophagitis is also seen in other reports from the Far East19, 20, 21, 22, 23 as well as from North America.24, 25, 26, 27, 28, 29, 30 The relationship, however, is not a simple one, and the effect of the infection on acid secretion is determined by a number of factors including the location and extent of the infection, the severity of the inflammation, which in part is determined by the strain of H. pylori, and the degree of gastric atrophy.
Figure 3: The odds ratio (95% confidence intervals) for prevalence of H. pylori in patients with esophagitis in North America24, 25, 26, 27, 28, 29, 30 and the Far East.19, 20, 21, 22, 23
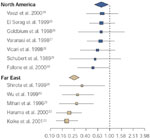
The large diamonds represent the summary odds ratio for each geographic area. The largest boxes represent studies with large sample sizes. (Source: Pandolfino et al.6, with permission from Blackwell Publishers.)
Antral gastritis, often associated with duodenal ulcer disease,31 can be associated in fact with increased acid secretion resulting from suppression of somatostatin release and therefore a reduction in its ability to inhibit gastrin release. Gastrin levels, therefore, increase.32 On the other hand, corpus gastritis results in decreased acid secretion largely owing to inflammatory damage to the parietal cells.33 The effect is compounded if gastric atrophy results.34 Commonly, the infection and inflammation tend to be more generalized and the effect to reduce acid secretion more prominent when the corpus is involved. Corpus gastritis is associated with a 54% reduced risk of esophagitis.25
The CagA, iceA1, and vac s1 types of H. pylori generally are more virulent strains, produce a more severe gastritis, are more commonly associated with ulcer disease, and are less commonly seen in GERD patients.35, 36, 37 This inverse relationship of GERD with more virulent strains of H. pylori may not be evident with milder degrees of esophagitis.38 Nevertheless, the bulk of evidence indicates that reduced acid secretion such as in the presence of H. pylori infection of the stomach is associated with a decreased prevalence of esophagitis. The protective effect also appears to apply to decreasing complications of GERD such as Barrett's esophagus.18
The efficacy of reducing acid secretion in the treatment of esophagitis, and the potential protective effect of reduced acid secretion with H. pylori infection, has led to some concerns. Would the treatment of H. pylori infection with an increase of acid secretion to at least normal levels lead to an increase in the incidence of esophagitis, and to a poor response to acid-suppression therapy of esophagitis or to higher recurrence rates after treatment? Presumably these adverse results would occur in those cases where more severe extensive gastritis involving the gastric corpus was reversed by treatment of the H. pylori.
Generally, treatment of H. pylori has not resulted in an increase in GERD in patients with gastric or duodenal ulcer unless other risk factors are present.8, 11, 39 Studies that include other types of subjects indicate that H. pylori treatment may increase acid secretion and the prevalence of mild esophagitis, more commonly if corpus gastritis was present or if other factors that predispose to reflux such as a hiatus hernia were also present.13, 23, 40
The presence of H. pylori has no significant effect on the successful treatment of reflux esophagitis with a proton pump inhibitor.15, 41, 42 The consensus is that eradication of H. pylori infection does not compromise acid-suppression treatment of esophagitis, and is not associated with an increased incidence of esophagitis recurrence with treatment of the esophagitis.8, 14, 43, 44 The relapse rate may be reduced, presumably if the infection has caused significant gastric atrophy and decreased acid secretion.41 Therefore, at present, there is no convincing evidence that the presence of H. pylori infection with its effect to reduce gastric acid secretion should preclude eradication of the infection because of concerns related to GERD or its treatment. Other factors are more important in determining the "acid burden" that produces esophagitis or other manifestations of GERD. Further, eradication of H. pylori has other potential benefits including removing a major risk factor for ulcer disease and a link to development of gastric cancer.
Other Refluxed Substances
The reflux of gastric content also includes pepsin and substances such as bile and pancreatic and intestinal enzymes from the duodenum. Although pepsin is activated at a pH <4, and the combination of acid plus pepsin is potentially more injurious to esophageal mucosa than acid alone,45, 46 the levels of pepsin in gastric juice and the maximum output of pepsin are not different in patients with or without esophagitis.16 That is, an acid pH is required for the deleterious effects of pepsin to become active. However, in this regard, the presence and amount of gastric acid are still of prime importance.
The role of duodenogastroesophageal reflux still remains controversial, although newer measurement techniques that measure bile content and assess the presence and movement of refluxed liquid in the esophagus are providing further insights. The presence of duodenogastroesophageal reflux alone as measured by bilirubin content did not produce esophagitis in partial gastrectomy patients. Patients with both acid and duodenal content in the esophagus had a high frequency (67%) of esophagitis,47 and duodenogastric reflux is more common in GERD patients with stricture or Barrett's esophagus.48 The frequency of esophageal exposure to both acid and duodenal content increases, acid and bile in tandem, and is highest in patients with complicated Barrett's esophagus.5 Therefore, as with pepsin, the presence of acid in the gastroesophageal refluxate is required for the duodenal content to have its potential deleterious effect on the production of esophagitis.
Whether duodenogastroesophageal reflux is important in the production of NERD is also still controversial.49 Perfusion of bile acids into the esophagus of humans can cause pain.50 Multichannel intraluminal impedence techniques, often combined with pH and bilirubin measurements, have started to explore this issue in normal subjects as well as in patients with GERD.4, 51, 52 It is clear that when acid is suppressed, gastroesophageal reflux of nonacid content still occurs and can be readily measured with impedence techniques, and can be associated with symptoms.53, 54, 55 The importance of these findings awaits further study.
Antireflux Mechanisms at the Gastroesophageal Region
The antireflux mechanism has at least two "sphincter" mechanisms: the intrinsic muscular sphincter known as the lower esophageal sphincter (LES), and the diaphragm that functions as an external sphincter-like mechanism.56 The presence of a hiatus hernia impacts unfavorably on both of these sphincter mechanisms. Other structures such as the phrenoesophageal ligament help maintain the anatomic integrity of the region, but their function otherwise is not established.57
The Lower Esophageal Sphincter
Normal Physiology
The LES is tonically closed at rest, maintaining an average pressure of about 20 mmHg, and serves to prevent gastroesophageal reflux. The circumferential profile is asymmetrical with the higher pressures in the left lateral portion of the sphincter.58, 59 To allow passage of a bolus, LES pressure falls within 1.5 to 2.5 seconds of a swallow and remains low for 6 to 8 seconds as the peristaltic contraction transverses the esophageal body. Two main peripheral neurons mediate active contraction and relaxation of the LES, acetylcholine being the excitatory neurotransmitter and nitric oxide the main inhibitory neurotransmitter. Relaxation of the LES can also in part occur when tonic vagal cholinergic excitation to the LES is turned off with a swallow.60
The intrinsic LES in the humans is composed of at least two muscles. The circular muscle forms only a partial ring (or semicircular clasp), and the gastric sling muscle runs on the left lateral aspect to complete this portion of the sphincter (Figure 4).61 The LES circular clasp and sling muscles are functionally different in many ways, each with unique contractile properties. The circular clasp muscle has significant spontaneous tone, whereas the sling muscle has little tone and is more responsive to cholinergic stimulation.62, 63, 64 The higher pressure in the left lateral position of the sling is reduced by atropine, whereas the pressure in the remainder of the LES circumference is unchanged by cholinergic blockade.62, 63 Furthermore, these two muscles demonstrate differences in resting membrane potential and voltage-gated K+ ion channel densities,65 and in the L-type Ca2+ channel and calcium handling.66, 67 For example, although influx of extracellular calcium is central to the maintenance of myogenic tone and acetylcholine (ACh)-induced contractility in both LES muscles, this influx occurs through an L-type Ca2+ channel in LES circular muscle, and a nifedipine-insensitive, non–L-type Ca2+ channel in sling muscle. It is not clear whether the circular muscle of the very distal esophageal body also contributes as part of the proximal LES.
Figure 4: The structure of the lower esophageal sphincter (LES).
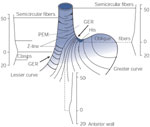
GER, gastroesophageal reflux. (Source: Liebermann-Meffert D et al.61, with permission from American Gastroenterological Association.)
These regional differences in the LES dictate that the mechanisms maintaining resting LES tone will be different for the LES smooth muscles—cholinergic excitation for the sling, and intrinsic myogenic tone for the circular clasp muscle—and set the basal conditions for the mechanisms necessary for LES relaxation. The circular clasp muscle with its high resting intrinsic tone is relaxed predominantly by release of nitric oxide (NO). There is little effect of NO on the sling muscle,68 and relaxation of the sling muscle is likely due predominantly to turning off its cholinergic excitation. That is, the dominant innervation of the circular clasp muscle is nitrergic and inhibitory, whereas that of the sling is cholinergic and excitatory.
The presence and location of the sling muscle has also been linked to maintenance of the acute angle of His at the greater curve aspect of the gastroesophageal junction, and the formation of a flap valve function that could also serve as an antireflux mechanism.69 This aspect remains controversial.
Pathophysiology: Low LES Pressure and TLESR
Gastroesophageal reflux occurs when LES pressure is lower than intragastric pressure. Under normal circumstances a small amount of reflux occurs when LES pressures are low with a swallow and during TLESR, and when increases in intraabdominal pressure or intragastric pressure overcome the resting LES pressure. The TLESR is independent of a swallow. Transient relaxation of the sphincter that is independent of a swallow can also occur with pharyngeal stimulation. However, these relaxations are different from the regular TLESR and do not appear to be associated with reflux. Their relaxation is less pronounced and they do not involve inhibition of the diaphragm.70, 71, 72
The nature of the LES changes and function associated with abnormal gastroesophageal reflux in the usual patients with GERD are well described and in large part determine the patterns, timing, and degree of reflux. In both patients and normal subjects, the TLESR in the face of a normal LES pressure is the most common mechanism of reflux, accounting for virtually all episode in normals and about two thirds in GERD patients.73, 74 Transient LES relaxation occurs largely in the postprandial period,73, 75 the increasing frequency of TLESR after a meal attributed to gastric distention.76 Not surprisingly and particularly in GERD patients, reflux with increases in intraabdominal pressure and free reflux through an abnormal low-pressure sphincter make up the remainder of the episodes.73
The resting pressure varies considerably throughout the day, and even normal physiologic events and daily activities provide times when LES pressures are lower and increase the potential for gastroesophageal reflux. The LES pressure increases in the recumbent position.77, 78 Fasting, pressures are higher during phase III of the migrating motor complex (MMC) and lowest during phase I.79 Feeding is often associated with a drop in LES pressure, resulting in large part from the secretion of hormones such as secretin and cholecystokinin (CCK) with fat intake,80, 81 or from the nature of the food itself or its contents, such as with chocolate,82 alcohol,83 and caffeine.84 Even colonic fermentation can lower LES pressure, the mechanism being unclear.85 Smoking decreases LES pressure,86 as does pregnancy, the latter due in part to the hormone progesterone.87 Many other hormones, neurotransmitters, and ingested medications can alter LES pressure, and those that do, such as anticholinergic drugs, nitrates, calcium channel blockers, and certain prostaglandins, can potentially predispose to gastroesophageal reflux.
The increased acid load in GERD patients and its relationship to low LES pressure and the severity of the esophagitis is also reflected in the pattern of reflux. The severity of GERD increases progressively from postprandial to upright, to supine, to bipositional reflux. A structural defect as reflected by decreased LES pressure and length is also significantly more common with supine and bipositional reflux88 (Figure 5). Therefore, reflux that occurs at night as well as during the day is particularly bad. That is, in addition to the absence of the beneficial effects of gravity, many of the physiologic changes that occur with sleep also favor development of GERD.89, 90 This aspect is discussed below.
Figure 5: Body position and gastroesophageal reflux causing mucosal injury.
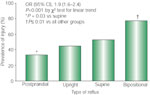
OR, odds ratio; CI, confidence interval. (Source: Campos et al.88, with permission from American Medical Association )
Low LES pressure and abnormal reflux can also occur in certain diseases, most commonly with the collagen vascular disorders such as scleroderma, where damage to the muscle and to the excitatory cholinergic innervation occur.91
The acid load to the esophagus is greater in GERD patients, in part because more TLESRs are associated with reflux even though the number of TLESRs is more or less similar.74, 92 In addition, the duration of the relaxation with a TLESR is longer than that with a swallow.93 The physiologic reason for the increased frequency of TLESR-associated reflux in GERD patients is unknown. The increase is not associated with changes in gastric compliance or meal-induced gastric relaxation in reflux patients.94 The mechanism likely relates to other factors such as the presence of a hiatus hernia.92, 95 In certain disorders, GERD may be associated with an increased frequency of TLESR as well, such as in obesity owing perhaps to increased sensitivity to distention,96 and in diabetes owing to the effect of hyperglycemia.97 Of particular interest is the effect of atropine. Although atropine decreases LES pressure, the frequency of TLESR is decreased and gastroesophageal reflux in fact decreases in both normal subjects and patients with GERD.98, 99 A central mechanism has been proposed for the decrease in TLESR,100 although atropine could potentially act peripherally as well by blocking the muscarinic-1 receptor that is present on the inhibitory nitrergic neuron of the LES.101
Pathophysiology: Differences in LES Circular and Gastric Sling Muscles
Knowledge of the regional clasp and sling differences has obvious clinical and therapeutic implications. However, virtually all of the clinical studies to date have not considered this aspect. Similarly, the studies of the effect of inflammation on the LES in vivo or in vitro have been directed only at the circular muscle. Nevertheless, attention to these differences holds the potential to give new insights for a better understanding of the pathogenesis of LES disorders, and consequently the rationale for old and new directions for therapy of these disorders. For example, in patients with gastroesophageal reflux the sling contraction could be augmented by pharmacologic manipulation of its cholinergic control, and gives credence to the use of cholinergic agonists in raising LES pressure for treatment of gastroesophageal reflux.102 Similarly, the surgical management of reflux could be tailored to the sling or circular muscle depending on careful three-dimensional pressure imaging of the LES in GERD patients.59 Significant attention has been directed to pharmacologic manipulation of the nitrergic control of the TLESR in patients with reflux.103, 104 As noted above, the dominant innervation of the circular muscle is nitrergic and inhibitory, whereas that of the sling is cholinergic and excitatory. Therefore, manipulation of the TLESR presumably is directed primarily at the circular muscle, although the role of the sling in the TLESR is unknown. The sling may also be involved through turning off of excitation or activation of its neural inhibition.
The additional role of the sling in maintaining an acute angle at the gastroesophageal junction remains controversial. An increase in the angle following partial gastrectomy is associated with increased reflux and reflux symptoms.105 In the presence of a hiatus hernia, the radial asymmetry of the LES pressure profile disappears, in part owing to removal of the external diaphragmatic sphincter.106 Presumably the sling also loses some of its sphincter function through either the anatomic distortion produced by the hernia or the effect of inflammation such as carditis and esophagitis on the muscle. This aspect requires further study.
The Diaphragm
Normal Physiology
The distal esophagus passes through the diaphragmatic hiatus that is usually formed by the right crus of the diaphragm. As a result the diaphragm can function as an "external sphincter" by compressing the gastroesophageal junction; this compression increases LES pressure by 10 to 100 mmHg with each inspiration, depending on the intensity of the inspiration.107, 108 Increases in intraabdominal pressure also increase LES pressure, in part through contraction of the diaphragm.109 The crural portion of the diaphragm relaxes with a swallow and the TLESR, and with belching and vomiting. The relaxation of the crural portion during the TLESR enhances the potential for gastroesophageal reflux, especially if LES pressure is low.
In the presence of a hiatus hernia, the diaphragmatic sphincter is distanced from the gastroesophageal junction and therefore loses its ability to function as an antireflux mechanism.
Phrenoesophageal Ligament
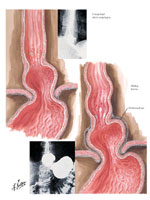
The phrenoesophageal ligament is a distinct structure that attaches the gastroesophageal junction to the diaphragm. It is stretched in hiatal hernia (Figure 6) (Figure 7). Its relationship to GERD has been the subject of controversy over the years.110, 111, 112 Nevertheless, its structure and placement provide good arguments for future studies to examine its functional role, and the nature of antireflux surgery that would take this role into consideration.57
Figure 6: The phrenoesophageal ligament at the gastroesophageal junction in normal and hiatal hernia.
(Source: Netter medical illustration used with permission of Elsevier. All rights reserved.)
Figure 7: Attachment of phrenoesophageal ligand to lower esophageal sphincter.
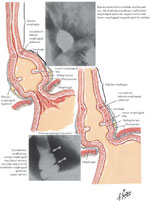
Note that the upper attachment of the ligament is at the level of the muscular ring (A ring) and the lower attachment is at the level of the mucosal ring (Schatzi, B ring) (Source: Netter medical illustration used with permission of Elsevier. All rights reserved.)
The "ligament" has two layers, a thin layer arising from the endothoracic fascia and a thicker circumferential continuation of the transversalis (endoabdominal) fascia. Before entering the esophageal wall, two leaves are formed. A thinner lower leaf runs caudally and attaches to the esophageal wall just above the angle of His, whereas the upper thicker leaf runs cranially to fuse with the esophageal adventitia above the diaphragm. The ligament is composed of abundant collagen and elastic fibers and some interspersed smooth muscle cells. The upper leaf is firmly attached to the esophagus with collagenous extensions that penetrate to the level of the submucosa, whereas the lower leaf only blends with the adventitia. The leaves become somewhat attenuated with age—the lower leaf more so, and this leaf can disappear.
The structure and attachment of the ligament provide a logical basis for its potential function. Its elastic properties and firm attachment superiorly would serve to limit displacement of the esophagus into the thorax and draw it back into position, while minimizing circumferential traction on the LES region. Attenuation of the ligament with age would facilitate the development of hiatus hernia.
Hiatus Hernia
Some notes on hiatus hernia and its relationship to GERD are made above. In brief, (1) reflux is facilitated by the presence of a hiatus hernia, the reflux increasing as hernia size increases.92, 95, 113, 114, 115, 116 (2) The LES pressure is decreased, and the compliance of the LES and the cross-sectional area are increased in GERD patients, and are greatest in those with hiatus hernia.106, 117 (3) The combination of a low-pressure sphincter and hiatus hernia is most common in patients with severe esophagitis and its complications, although the presence of a hiatus hernia alone also signals the presence of increased reflux and more severe esophagitis.115, 118 (4) Esophageal clearance of acid is decreased in the presence of a hiatus hernia.119, 120
The mechanisms whereby the hiatus hernia is related to a decreased LES pressure, decreased acid clearance, and increased reflux are multiple and not fully understood: (1) The low LES pressure results in part from removal of the external diaphragmatic sphincter,106 and in addition from inflammatory effects on the intrinsic sphincter muscles and their innervation (see below). (2) Although the physiologic reason for the increased proportion of TLESR that are associated with reflux in GERD patients is unknown, the mechanism may relate to the presence of a hiatus hernia.92, 95 The frequency and the number of common cavity phenomena of TLESR decrease after a fundoplication antireflux repair, perhaps owing to alteration of the fundal and vagal afferent mechanisms that induce TLESR.121, 122, 123 (3) The hernia acts as a reservoir, the diaphragm trapping acid therein, allowing for re-reflux of acid and perhaps increased tendency to inflammation at the cardia (carditis).124, 125 (5) Stretch from the lower arm of the phrenoesophageal ligament could potentially put circumferential traction on the LES, especially in younger patients before degeneration of the ligament occurs with aging.57
Mucosal Defense Mechanisms and Inflammation
With gastroesophageal reflux, a greater duration of exposure of the esophagus to a pH <4 leads to increasing severity of esophagitis and its complications (Figure 7).5, 48 Both the LES mechanisms that produce excess reflux of acid and the esophageal body motility disorders that result in prolonged acid exposure to the mucosa contribute to the duration of the exposure.
Inflammation: LES and Esophageal Motility
As noted above, some diseases such as scleroderma can lead to GERD through damage to neural control and muscle resulting in hypomotility of the LES and esophageal body. More commonly, the presence of inflammation of the mucosa and esophageal wall is associated with decreased LES pressure and esophageal motility that can impact on the acid burden to the mucosa through increased reflux and delayed esophageal clearance.126 In the body, both decreased amplitude of the primary and secondary peristaltic waves, and failed peristalsis are common. Irregular contractions can also occur.20, 118, 126, 127, 128, 129, 130 A vicious cycle results (Figure 2). Both a low LES pressure and disturbed esophageal motility are more common and pronounced in patients with more severe esophagitis, but because of the vicious cycle it is not clear which is the cart and which the horse.
Nevertheless, evidence is accumulating that inflammation affects nerves and muscle to alter LES and esophageal body motility. Both a decrease in cholinergic excitatory and an increase in nitrergic and other inhibitory mechanisms appear to be involved. This combination would result in hypomotility. The studies have involved only the circular muscle and not the sling.
The decrease in cholinergic excitation may have a vagal component.131 Animal experiments and studies of human LES tissue demonstrate a decrease in local cholinergic excitation,132, 133, 134 and major changes in calcium stores135 and in the intracellular pathways that mediate tone, and excitation contraction coupling in response to acetylcholine (ACh).136 In particular, prostanoids are involved. Inflammatory mediators such as interleukin-1B (IL-1B) and increases of reactive oxygen species (e.g. H2O2) are associated with increases in prostaglandin E2 (PGE2) and an isoform of PGE2. Prostaglandin E2 relaxes the LES, whereas the isoform of PGE2 blocks prostaglandin F2a (PGF2a)-mediated contraction.137 Prostaglandin E2a along with thromboxane A2/B2 are important in maintenance of LES tone, and blockade of PGE2a activity further reduces LES tone.138 Recent studies also indicate that inflammation induces the production of IL-6 in the mucosa and that IL-6, but not (IL-1B), leads to an increase of H2O2 in the muscle.139 The H2O2 appears to be the main culprit that causes increases in platelet-activating factor (PAF) and PGE2, both of which can act to reduce both ACh release140 and LES muscle tone.141 Earlier studies indicated that inhibition of prostaglandin synthesis with indomethacin prevented or corrected esophagitis-associated LES hypotension, presumably through a reduction of PGE2.142 Esophageal IL-8 is also increased in reflux esophagitis, and presumably enhances neutrophil trafficking.143
In addition to prostanoid effects, inflammation is associated with increased NO in esophageal tissues134, 144 and evidence of increased activity of the nitrergic inhibitory innervation.132, 134 These changes also result in low LES pressure and decreased esophageal body motility.
Of interest, acid infusion causes shortening of the esophagus145 in response in part to inflammatory mediators,146 and NO contracts longitudinal esophageal smooth muscle.147 These responses to acid and acid-induced inflammation have been proposed as potential factors contributing to the development of hiatus hernia.
Sensory Mechanisms and Symptoms
Although acid or inflammatory mediators are accepted as putative initiators of esophageal symptoms, the mechanisms whereby these gain access to sensory pathways and subsequently produce symptoms either directly or through other events such as abnormal muscle contraction remain the subject of controversy, speculation, and research.49, 148, 149 A number of issues remain in this area: (1) Why do patients experience heartburn in the absence of abnormal acid exposure or esophagitis (NERD)?150, 151 (2) Why do some patients with severe esophagitis and complications such as Barrett's esophagus have absent or little heartburn?152 (3) Is visceral hypersensitivity peripheral and/or central, hyperalgesia or allodynia?152, 153, 154, 155, 156 (4) Do sensations such as heartburn arise from abnormal contractions such as sustained esophageal contractions of circular or longitudinal muscle, and/or distention,156, 157, 158 and is more proximal reflux important?2, 4 (5) Is mucosal dysfunction that produces abnormal tissue resistance an explanation for some or all of the controversy?148 (6) Do the age and sex of the patients play a role?159, 160, 161 (7) To what extent is NERD a "functional" disorder" of the esophagus?162
Other Factors
Gastric emptying. Gastric emptying as determined recently by a standardized technique confirms that delayed emptying is common in GERD patients, present in 26% of patients on the basis of retention at 4 hours.163 There are cogent reasons to suppose that delayed emptying could lead to GERD, including increased gastric content that could increase the frequency of TLESR and gastric acid secretion.164 However, there is little evidence to support this conclusion, as most patients do not have delayed gastric emptying and the delay is probably only an enhancing cofactor in a minority of patients. In fact, in some patents with foregut symptoms, delayed gastric emptying may be associated with decreased postprandial reflux.165 Similarly, in children there is no definite relationship of delayed gastric emptying to GERD unless other abnormality such as malrotation of the stomach is present.166, 167
Saliva and acid neutralization. Acid neutralization by swallowed saliva serves to "clear" the remaining acid after the bolus is emptied from the esophagus, and does so in a stepwise fashion with each swallow-induced peristaltic contraction.168 Acid secretion increases in both normal subjects and patients with reflux esophagitis,169 the increase related primarily to the appearance of heartburn.170 There is no evidence that a defect in salivary flow or acid neutralizing capacity contributes to the development of GERD in the usual patient. In diseases associated with the sicca complex there is no increase in reflux symptoms,171 although in some such patients decreased salivary flow may be associated with esophageal injury.172
Sleep. Although the frequency of reflux episodes decreases during sleep,75, 173 many of the physiologic changes that occur with sleep favor development of GERD.89, 90 These changes include (1) increased acid secretion that can also be seen as "nocturnal breakthrough" in GERD patients on proton pump therapy174, 175, 176; (2) a marked decrease in acid clearance with enhanced proximal migration owing to loss of gravitational effects in the supine position,177, 178 decreased frequency of swallowing and therefore of primary peristalsis,179, 180 decreased saliva production,181 decreased perception of heartburn89; and (3) changes in the gastric slow-wave electrical activity182 that could predispose to decreased gastric emptying.183 Other factors such as sleep apnea and its relationship to obesity are also linked to GERD.89, 184 The mechanism for this relationship is unclear and may relate to apnea-related changes in intrathoracic pressure, or to obesity-related factors such as increased intraabdominal pressure and hiatus hernia.185, 186
Obesity. The relationship of obesity to GERD is controversial. Obesity has been found to be a risk factor for GERD,187 and weight loss may188 or may not189 be associated with improvement in GERD symptoms. Another study has found no relationship of obesity with GERD symptoms.190 Any relationship may in large part be determined by the presence of hiatus hernia in obese patients.186
Genetics. There is considerable evidence that there exists an inherited tendency to develop GERD.191, 192, 193 The evidence has derived largely from epidemiologic and family studies194, 195, 196, 197 and twin studies.198, 199 Many issues remain unresolved and the stimulus for continuing research: (1) Pediatric and adult GERD have both similar and different clinical features that will impact on genetic studies.200 (2) There may be a predetermined genetic predisposition to develop different phenotypes of GERD (NERD, erosive esophagitis, and Barrett's esophagus).192 This issue is particularly relevant to the relationship of GERD to adenocarcinoma of the esophagus.197, 201 (3) Some of the genetic predisposition to develop GERD likely rests in the risk factors such as obesity and hiatus hernia.202, 203 (4) Chromosomal determinants demonstrate a multitude of identifiable relationships because of the many factors involved in the pathogenesis of GERD. For example, mapping of a gene on chromosome 13q14 has been found in some patients204 but not in others.205
Conclusion
Gastroesophageal reflux disease has a multifactorial pathogenesis. Therefore, future studies must explore the many factors involved. A number of factors that impact on the mechanisms regulating competence of the gastroesophageal junction are of particular importance: (1) Structure and function of the phrenoesophageal ligament: its degeneration with age may predispose to development of hiatus hernia. Should surgeons take another look at reconstituting this structure? (2) Many agents that either increase or decrease the TLESR through central or peripheral action have already been addressed. Studies should take a closer look at the anticholinergic drugs, particularly M1 antagonists that could act at the peripheral nitrergic inhibitory neuron. (3) The marked regional differences in the properties of the LES circular clasp and sling muscle components hold the potential to explore differential abnormalities of these in GERD, and the potential for new medical and surgical approaches to therapy. Further study is needed to determine the relationship of the distal esophageal body circular smooth muscle to the LES and GERD. (4) The sensory and cognitive-behavioral aspects of patients with NERD require careful study to link the physiologic and psychological aspects, as has been done with patients with other disorders that have a major "functional" component such as nonulcer dyspepsia and irritable bowel syndrome.
