
Abstract
Purpose: Autosomal dominant CHARGE syndrome (OMIM no. 214800) is characterized by choanal atresia or cleft lip or palate, ocular colobomas, cardiovascular malformations, retardation of growth, ear anomalies, and deafness, and is caused by mutations in the CHD7 gene. Here, we describe the outcome of a molecular genetic analysis in 18 Finnish and 56 German patients referred for molecular confirmation of the clinical diagnosis of suspected CHARGE syndrome.
Methods: Quantitative real-time polymerase chain reaction or multiplex ligation-dependent probe amplification assays did not reveal deletions in mutation negative cases, suggesting that larger CHD7 deletions are not a major cause of CHARGE syndrome.
Results: In this group of 74 patients, we found mutations in 30 cases. 22 mutations were novel, including 11 frameshift, 5 nonsense, 3 splice-site, and 3 missense mutations. One de novo frameshift mutation was found in the last exon and is expected to result in a minimally shortened CHD7 polypeptide. Because the mutation is associated with a typical CHARGE syndrome phenotype, it may indicate the presence of an as yet unknown functional domain in the very carboxyterminal end of CHD7.
Conclusions: Our mutation detection rate of 40.5% is reflective of screening an unselected sample population referred for CHD7 testing based on suspected clinical diagnosis of CHARGE syndrome and not for having met strict clinical criteria for this disorder.
Similar content being viewed by others


Main
CHARGE syndrome (MIM no. 214800) is an autosomal dominant congenital malformation disorder characterized by variable occurrence of choanal atresia or cleft lip or palate, ocular colobomas, cardiovascular malformations, retardation of growth, ear anomalies, and deafness.1–4 Its incidence varies from 1:8500 to 1:10,000.1,5 The current clinical criteria for the diagnosis of CHARGE syndrome were defined by Blake et al. in 19981 and further updated by Verloes in 2005.6 Patients likely to have CHARGE syndrome harbor either four major symptoms (choanal atresia, coloboma, characteristic ears, and cranial nerve anomalies) or three major and three of the minor symptoms including cardiovascular malformations, genital hypoplasia, cleft lip/palate, tracheoesophageal fistula, growth deficiency, developmental delay, and distinctive facial features.
The molecular etiology of CHARGE syndrome was revealed in 2004 by Vissers et al.7 They reported two patients who had overlapping microdeletions in 8q12 region spanning the CHD7 gene (chromodomain helicase DNA-binding protein 7). By subsequent sequencing of this gene, heterozygous intragenic mutations were found in 10 of 17 patients. Further studies have confirmed mutations in CHD7 gene as the major cause of CHARGE syndrome.7–14 Most of the mutations are unique and occur de novo, but a few cases with germline mosaicism and familial inheritance have been reported.11,12 No evidence of genotype-phenotype correlation has been observed even in large cohorts of patients, and, for example, monozygotic twins with an identical mutation have been reported to present variable phenotypes.11,12 In some patients, phenotypes show overlapping features with those described in other syndromes, such as velocardiofacial or DiGeorge and Kallmann syndromes.13 Also, genetic heterogeneity has been reported in patients with a clinical diagnosis of CHARGE syndrome. Devriendt et al.15 have described a patient with features of CHARGE phenotype and a submicroscopic deletion in chromosomal area 22q11.2, and a missense mutation in another gene, SEMA3E, has been described in one patient.16
The CHD7 protein belongs to a family of highly conserved proteins involved in regulation of transcription and it has several functional domains including HELICc, SNF2-related helicase/ATPase, BRK, and chromodomains.17 Most likely, it plays a significant role in early embryonic development and controls gene expression through its activity in chromatin remodeling. Lalani et al.12 studied the expression pattern of the gene in mouse embryos and observed markedly variable relative expression levels in different tissues. The highest expression levels were in the tissues in which congenital abnormalities are frequently found in CHARGE syndrome. Sanlaville et al.14 studied the gene expression in 10 human fetuses with truncating CHD7 mutations and similarly, observed the correlation between the expression pattern and congenital malformations.
To date, more than a hundred different point mutations have been described throughout the CHD7 gene. Despite the known molecular etiology, confirmation of the clinical diagnosis of CHARGE syndrome may not be straightforward. The large size of the gene and the variability of the clinical features present a challenge in developing a molecular diagnostic protocol. In the present survey, we have analyzed a series of samples of individuals with features suggesting CHARGE syndrome to confirm the clinical diagnosis and report the identification of new and recurrent mutations in the CHD7 gene.
MATERIALS, PATIENTS, AND METHODS
Patients
Blood and tissue samples from 74 patients, 18 in Finland and 56 in Germany, were referred to our laboratories for clinical screening of mutations in the CHD7 gene to confirm the clinical diagnosis of CHARGE syndrome. Informed consent was obtained from all human subjects before analysis. There were no preselection criteria other than clinical evaluation according to the expertise of the referring clinicians. At the time of diagnosis, age of the patients varied from 23 days to 32 years. Three of the patients had died of complications in infancy, and in one case, the pregnancy was prematurely terminated due to malformations detected in the ultrasound scan. Whenever available, parental samples were also studied to confirm the de novo occurrence of the mutations.
Genetic analysis
DNA was extracted from peripheral blood lymphocytes, fibroblasts, and liver cells according to standard protocols. Primers covering exons 2–38 and the exon-intron boundaries of the CHD7 gene (GenBank accession NM_017780) were designed with the Primer3 software.18 Polymerase chain reactions (PCRs) were performed in a standard 25 μL reaction volume with 100 ng of template DNA. For sequencing, PCR products were purified with an ExoSAP enzyme mix (USB Corporation, Cleveland, OH). All exons were sequenced in both directions using the BigDye Terminator v3.1 cycle sequencing kit and ABI Prism 3100/3130 DNA sequencers (Applied Biosystems, Foster City, CA). Novel missense mutations were screened in 200 ethnically matched control chromosomes.
Multiplex ligation-dependent probe amplification
In the Finnish laboratory, deletions encompassing the CHD7 gene were excluded with multiplex ligation-dependent probe amplification (MLPA). The analysis of the CHD7 gene was performed with P201 SALSA kit (MRC-Holland, The Netherlands). The reactions were performed in 0.25-fold volume of the reagents and 3-hour hybridization modified from the manufacturer's protocol with 100 ng of genomic DNA. The MLPA PCR products were separated with ABI 310 capillary DNA sequencer (Applied Biosystems). The chromatograms were analyzed with the GeneMarker software (SoftGenetics, State College, PA). For each sample, relative probe signals were defined by dividing each peak area measured by the area of the combined control peak areas in that sample, and relative values were compared to those of control samples.
Quantitative real-time PCR
In the German laboratory, to exclude deletions encompassing the CHD7 gene quantitative real-time PCR was performed with 11 intragenic amplicons in exons 2, 7, 13, 15, 20, 22, 30, 34, 35, 36 and in intron 5. Primer design and experimental procedure followed the approach reported by the German laboratory for deletion screening in the gene SALL4.19 In the Finnish laboratory, to confirm the results of MLPA analysis, primers were designed for exon 31 in the gene CHD7 and for exon 8 in the control gene GAPDH with the Beacon Designer software (PREMIER Biosoft International, Palo Alto, CA) to amplify 159-bp and 162-bp fragments, respectively. Reactions were performed in triplicates using 12.5 ng DNA in a 25 μL reaction with 1-fold iQ SYBRGreen supermix and the iCycler thermal cycler (Bio-Rad, Hercules, CA). Fourfold dilutions ranging from 25 to 0.0976 ng were used to prepare the standard curve, and the relative difference in the copy number ratio of CHD7 and GAPDH between the control and patient samples was analyzed with the Pfaffl method.20
mRNA analysis
To study the effects of the pathogenic role of the splice-site mutations at RNA level, patient- and control-derived phytohemagglutinin-stimulated and Epstein-Barr virus-induced lymphocytes were cultured in RPMI medium, and the total RNA was extracted using an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Primers for the cDNA amplification were designed with Primer3 software. Reverse transcription and cDNA amplification were performed in a one-tube reaction with the Titan One Tube RT-PCR kit (Roche Diagnostics, Mannhein, Germany) using 100 ng of the total RNA. The amplified cDNA fragments were sequenced with the ABI 310 capillary DNA sequencer.
RESULTS
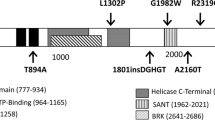
In our analysis of 74 patients, 30 (40.5%) intragenic mutations were identified (Table 1). Of these 30 mutations, 22 were novel and 8 have been published previously. The majority (20 of 30, 67%) were truncating mutations, either nonsense (9) or frameshift (11) mutations, causing a premature stop codon. The truncating mutations were scattered throughout the coding area indicating no mutational hotspot. One frameshift mutation, c.8962dupG (p.D2988fsX1), was located only 33 bp upstream from the 3′-end of the coding sequence. It occurred de novo and was therefore considered pathogenic. The nonsense mutation p.R2284X, previously published by Felix et al.,10 was identified in two unrelated patients in this study. Four splice-site mutations (4 of 30, 13%) were detected, three within consensus splice sites and one 17 bp upstream of the 5′-end of exon 26 (c.5405–17G→A). A mRNA analysis of this mutation revealed the formation of a cryptic splice site and, as a result, five codons were inserted into the cDNA in-frame. Six missense mutations (6/30, 20%) were found scattered throughout the entire length of the gene. Two mutations in the functional domains, p.I1028V in the SNF2-related helicase/ATPase domain and p.Q1395H in the HELICc domain, are highly likely to be pathogenic due to their destroying effect on the function on these domains. The p.I1028V mutation has previously been published in two studies.7,11
The analysis of parental samples revealed one familial mutation in our series. In family CH4, an affected father had transmitted nonsense mutation p.Q1599X to both of his children. Additional parental samples were available for analysis in 12 cases with truncating mutations, but, in all of these, the mutations had occurred de novo. For splice-site mutations, the parental analysis confirmed de novo occurrence in three of the four cases; in one case (c.5895–2A→G), parental samples were not available. Among the six missense mutations, p.I1028V and p.Q1395H were confirmed to have occurred de novo, and in a case of p.C1643W, the maternal sample was available but showed no mutation. In the case of three other missense mutations (p.W983G, p.D1596G, and p.R2319C), parental samples were not available. These three mutations were not detected in the 200 control chromosomes. In addition, the mutations p.D1596G and p.R2319C have been previously published.10,11 Deletions encompassing the CHD7 gene were not detected.
In addition to the putative pathogenic mutations, 10 new polymorphisms were detected in introns 5, 16, 22, 29 and in exons 2, 4, 31, 32, and 38 (Table 2). Of them five were missense changes found either in patients with a truncating mutation or in one unaffected parent.
DISCUSSION
Since the original article by Vissers et al. in 2004,7 mutations in the CHD7 gene in CHARGE syndrome patients have been found at a detection rate varying between 58% and 90%.7–14 In the present study, we have analyzed a series of 74 samples from patients referred to our diagnostic laboratories for confirmation of the clinical diagnosis of CHARGE syndrome. We identified 30 intragenic CHD7 mutations, corresponding to a 40.5% detection rate in this unselected group of patients. The failure to detect larger deletions in the mutation negative cases illustrates that larger deletions—as detected previously7—are not contributing substantially to the CHD7 mutational spectrum.
Most of the mutations (73%) found were novel. In accordance with previous reports, most were truncating mutations causing a premature stop codon and probably leading to haploinsufficiency of CHD7. Familial mutations in CHARGE syndrome are rare, but in our series, one nonsense mutation was shown to be familial because it was transmitted from an affected father to both of his children. The father is very mildly affected, he has characteristic ears, and his face is slightly asymmetrical, but his intelligence is normal, and he has no genital anomalies. Two of the six missense mutations were located in known functional domains of the polypeptide and can hence be considered pathogenic. In addition, two other missense mutations had previously been described in other studies, supporting their pathogenic role. Three of the four splice-site mutations are also likely to be deleterious due to exon skipping or formation of the premature stop codon. The precise effect was analyzed in one case, in which the mutation resulted in the formation of a cryptic splice site and insertion of five codons into the cDNA in-frame. This change possibly alters the three-dimensional structure of the CHD7 protein and thereby disturbs the normal function.
One mutation found in our series is particularly interesting. p.D2988fsX1 occurred de novo in a case fulfilling clinical criteria for CHARGE syndrome and is therefore considered to be pathogenic. Because it is a truncating mutation in the last exon of CHD7, the mutation is expected to escape nonsense-mediated mRNA decay. Only one aberrant amino acid is introduced into the truncated CHD7 protein, and therefore this mutation may indicate an as yet unknown functional domain in the most 3′ exon of CHD7.
In conclusion, our molecular genetic analysis of a cohort of patients with clinically diagnosed or suspected CHARGE syndrome revealed mutations in 40.5% of the patients. However, in most cases, sufficient clinical data were not available to evaluate whether the patients fulfilled the current clinical criteria by Blake and Prasad9 and Verloes.6 Samples were referred to us by several clinicians in two different countries, Finland and Germany, and it is likely that the criteria for the clinical diagnosis or suspicion of CHARGE syndrome differed. Obviously, the mutational detection rate depends on the criteria used when sending samples for diagnostic studies. However, we believe that this patient material represents well the samples sent to a routine diagnostic laboratory. The mutation detection rate of 40.5% (which was similar in both contributing institutions) clearly confirms the utility of molecular screening in CHARGE syndrome, and gives an estimate as to what to expect when referrals include cases fulfilling the diagnostic criteria as well as such which do not, but for which CHARGE syndrome is nevertheless one of the main differential diagnoses.
References
Blake KD, Davenport SL, Hall BD, Hefner MA, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998; 37: 159–173.
Hall BD . Choanal atresia and associated multiple anomalies. J Pediatr 1979; 95: 395–398.
Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ . Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation—a syndrome. J Pediatr Ophthalmol Strabismus 1979; 16: 122–128.
Pagon RA, Graham JM Jr, Zonana J, Yong SL . Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 1981; 99: 223–227.
Issekutz KA, Graham JM Jr, Prasad C, Smith IM, et al. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A 2005; 133: 309–317.
Verloes A . Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A 2005; 133: 306–308.
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004; 36: 955–957.
Aramaki M, Udaka T, Kosaki R, Makita Y, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr 2006; 148: 410–414.
Blake KD, Prasad C . CHARGE syndrome. Orphanet J Rare Dis 2006; 1: 34
Felix TM, Hanshaw BC, Mueller R, Bitoun P, et al. CHD7 gene and non-syndromic cleft lip and palate. Am J Med Genet A 2006; 140: 2110–2114.
Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet 2006; 43: 306–314.
Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet 2006; 78: 303–314.
Ogata T, Fujiwara I, Ogawa E, Sato N, et al. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr J 2006; 53: 741–743.
Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 2006; 43: 211–217.
Devriendt K, Swillen A, Fryns JP . Deletion in chromosome region 22q11 in a child with CHARGE association. Clin Genet 1998; 53: 408–410.
Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, et al. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 2004; 41: e94.
Woodage T, Basrai MA, Baxevanis AD, Hieter P, et al. Characterization of the CHD family of proteins. Proc Natl Acad Sci U S A 1997; 94: 11472–11477.
Rozen S, Skaletsky H . Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Borozdin W, Boehm D, Leipoldt M, Wilhelm C, et al. SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenetic mechanism. J Med Genet 2004; 41: e113.
Pfaffl MW . A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45.
Acknowledgements
Supported by the University of Turku Foundation, Special Federal Grant of Turku University Central Hospital.
We thank the patients and their families for their cooperation, the clinicians for referring patient samples for CHD7 testing, and Bernd Rösler and Tanja Velten for expert technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Vuorela, P., Ala-Mello, S., Saloranta, C. et al. Molecular analysis of the CHD7 gene in CHARGE syndrome: identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet Med 9, 690–694 (2007). https://doi.org/10.1097/GIM.0b013e318156e68e
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318156e68e
Keywords
This article is cited by
-
A New Model for Congenital Vestibular Disorders
Journal of the Association for Research in Otolaryngology (2019)
-
An unclassified variant of CHD7 activates a cryptic splice site in a patient with CHARGE syndrome
Human Genome Variation (2018)
-
Next-generation sequencing of patients with congenital anosmia
European Journal of Human Genetics (2017)
-
Rapid Discovery of De Novo Deleterious Mutations in Cattle Enhances the Value of Livestock as Model Species
Scientific Reports (2017)
-
Unique phenotype in a patient with CHARGE syndrome
International Journal of Pediatric Endocrinology (2011)
