
Abstract
Purpose: To provide a summary of the outcomes of two working conferences organized by the Centers for Disease Control and Prevention (CDC), to develop recommendations for practical, sustainable mechanisms to make quality control (QC) materials available to the genetic testing community.
Methods: Participants were selected to include experts in genetic testing and molecular diagnostics from professional organizations, government agencies, industry, laboratories, academic institutions, cell repositories, and proficiency testing (PT)/external Quality Assessment (EQA) programs. Current efforts to develop QC materials for genetic tests were reviewed; key issues and areas of need were identified; and workgroups were formed to address each area of need and to formulate recommendations and next steps.
Results: Recommendations were developed toward establishing a sustainable process to improve the availability of appropriate QC materials for genetic testing, with an emphasis on molecular genetic testing as an initial step.
Conclusions: Improving the availability of appropriate QC materials is of critical importance for assuring the quality of genetic testing, enhancing performance evaluation and PT/EQA programs, and facilitating new test development. To meet the needs of the rapidly expanding capacity of genetic testing in clinical and public health settings, a comprehensive, coordinated program should be developed. A Genetic Testing Quality Control Materials Program has therefore been established by CDC in March 2005 to serve these needs.
Similar content being viewed by others
INTRODUCTION
With the rapid growth of genetic testing in clinical and public health practice, assuring quality testing has been recognized as a major public need. In order to fulfill its public health mission of improving the quality of laboratory testing and practices, the Division of Laboratory Systems, under the Public Health Practice Program Office (now under the Coordinating Center for Health Information and Service) of CDC, initiated studies in 1999 through 2000, to 1) assess the status of quality assurance practices of laboratories performing genetic testing, 2) identify issues critical for establishing quality assurance and performance evaluation programs, and 3) develop recommendations for performance improvement in genetic testing. Of the five core recommendations generated from these studies, improving the availability of QC materials was identified as the issue of utmost urgency, based on the consensus that the lack of appropriate QC materials for many genetic tests is a key factor inhibiting the development of comprehensive quality assurance and performance evaluation programs for genetic testing.1,2
QC materials are essential for validating performance characteristics of new tests, monitoring test performance, and detecting errors in the testing process. Unlike other areas of laboratory testing for which diagnostic kits or testing systems containing QC samples are often available, FDA-approved, commercial diagnostic kits are presently available for only a few genetic tests;3 while the majority of genetic tests in current use have been developed in-house by individual laboratories. Laboratories obtain the needed control samples and other QC materials through a number of sources, including: 1) commercially available cell lines or DNA samples; 2) previously tested patient specimens; 3) interlaboratory exchanges; 4) colleagues and authors of relevant publications in clinical or research settings; and 5) materials from PT/EQA surveys, such as those available from the College of American Pathologists (CAP), the European Molecular Genetics Quality Network (EMQN), and other US or international EQA schemes.4 For most genetic tests, however, the desired appropriate QC materials are often difficult or impossible to obtain. This is especially true for tests evaluating heterogeneous mutations or complex variations, tests based on very recent gene discoveries, and tests for rare diseases or rare mutations. For example, the implementation of the recommendations of the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics (ACMG) for preconception and prenatal carrier screening for cystic fibrosis (CF) has made it the highest-volume genetic test in the U.S. since 2001.5 Although positive control samples for the majority of the recommended panel of mutations and variants8 and other sources, some alleles that are often included on laboratories' testing platforms became publicly available only recently as a result of a CDC-funded project to develop positive samples for molecular genetic testing.9,10
The lack of appropriate samples has been recognized as a barrier to interlaboratory performance evaluation for genetic testing since PT/EQA challenges need to represent mutations or situations that laboratories may encounter in practice. Furthermore, the availability of control materials often trails the rapid advances in genomic research and the adaptability of molecular diagnostic technologies, presenting a major challenge for the development and evaluation of new genetic tests.1
Multiple approaches have been undertaken to develop QC materials to meet the needs of the genetics community. In 2000, CDC competitively funded two collaborative projects to explore novel methods for accruing human mutation samples for molecular genetic testing. One of these projects, which was carried out by Dr. Timothy Stenzel and others in Duke University, demonstrated a process of using residual patient samples to establish stably transformed cell lines that can be used for QC and PT/EQA purposes.9 The other project was undertaken by Dr. Wayne Grody and others in the University of California at Los Angeles School of Medicine, to generate artificially constructed mutation samples that resemble natural mutation-containing human cell lines.11 Other government agencies have also engaged in efforts in this area. For example, the Coriell Cell Repositories, funded by the National Institute for General Medical Sciences (NIGMS), has been used by the genetics community as a major material resource.12 The National Institute of Standards and Technology (NIST) has made available standard reference materials (SRMs) for mitochondrial DNA analysis and for trinucleotide repeat measurement for the fragile X mental retardation 1 (FMR1) gene analysis.13,14 In addition, in light of the need for efficient control materials in multiplex or panel testing, efforts have been made to develop synthetic or artificially constructed controls.15,16 In the international community, improving material availability has been recognized as a priority as well. Both the CRMGEN project and a major component of the more recent Eurogentest project are funded by the European Commission to develop and make available reference materials for genetic tests.17,18
In order to expand the experience from current efforts, and more importantly, to establish a sustainable process to make QC materials available to the genetic testing community, CDC organized two working meetings in 2003 and 2004, respectively, with an emphasis on identifying areas of need for human molecular genetic testing and developing recommendations in this initial direction. For discussions at the working meetings that led to this report, QC materials referred to materials intended for use in control procedures, test validation, quality assurance, external quality assessment, and test development. Such materials may be described as “control materials,” “controls,” or “reference materials,” as defined by respective sources (Table 1).
METHODS
On September 15-16, 2003, CDC organized the first QC Materials for Genetic Testing meeting in Atlanta, GA. Participants of this working meeting included more than 50 leaders in genetic testing and molecular diagnostics from professional organizations, government agencies, industry, laboratories, cell repositories, and academic institutions. The main goals of this meeting were to 1) review the current efforts to develop QC materials for molecular genetic tests and 2) identify areas of need and issues that should be addressed to develop sustainable, practical mechanisms for providing QC materials to the genetic testing community.
During the meeting, panel discussion sessions were held to consider the ability of existing approaches to meet current and future needs for QC materials. Key issues that participants discussed included QC material needs at present and in the near future; strengths and weaknesses of current efforts; impact of patents and licensing agreements on development and distribution of QC materials; mechanisms for material development, collection, storage, validation, and distribution; provision of materials needed for genetic tests for rare diseases; cost considerations for obtaining validated QC materials; and models and mechanisms for a sustainable process.
At the conclusion of the meeting, participants identified the following eight areas of need for developing and providing QC materials for genetic tests: Assessment of current professional guidelines for appropriate use of QC materials to determine if further guidance is needed
-
Clarification of regulatory oversight for providers and users of QC materials
-
Development of a scheme for setting priorities for current and future needs
-
Review of current research activities on QC material development to facilitate collaboration and to identify further research needs
-
Development of networks of material contributors to facilitate participation and material collection
-
Development of approaches to using existing cell banks and repositories as material sources
-
Development of standardized processes for validation of QC materials
-
Development of better coordination to enhance support and resources needed for a sustainable process.
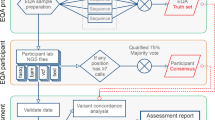
Recognizing the broad scope of these issues and the need to commit focused discussion on each, a total of eight workgroups were established, each addressing one of the identified needs. Figure 1 illustrates steps and factors involved in the process and the area of need that each workgroup focused on. Workgroup leaders were identified at the meeting and were charged to form their respective workgroups from meeting participants and additional experts with the expertise needed for the assigned task.

Steps and factors involved in the process to provide QC materials for genetic testing. Boxes with a folded corner indicate the areas of need that the QC materials workgroups addressed.
Each workgroup was provided with a list of specific issues that had been identified during the meeting (Table 2). Subsequently, each workgroup held teleconferences to discuss the issues and to develop approaches to addressing them.
During the second QC Materials for Genetic Testing meeting on March 8, 2004, in Orlando, Florida, the entire group met to review activities and progress of the eight workgroups; and develop recommendations and future directions for practical, sustainable mechanisms for making QC materials available to the genetic testing community. At this meeting, the workgroups held concurrent breakout discussions to review progress made to address each area of need and to develop further recommendations. During the subsequent general session, each workgroup presented a progress report that summarized their discussion and considerations. The reports were then discussed and commented upon by the entire group and other meeting participants.
RESULTS
The discussions at the meeting and the workgroup reports constituted the basis for identifying areas of need and developing recommendations, which then were commented on and enriched by other workgroups and participants at the meetings. The following is a summary of the key issues that the group deliberated upon and addressed.
Professional guidance on appropriate use of QC materials
To provide a landscape of professional guidance on use or inclusion of QC materials in genetic tests, the Professional Guidance Workgroup conducted a review of current U.S. and international practice guidelines and best practice recommendations for genetic testing, including general and disease-specific guidelines from professional societies and standard-setting organizations, publications by leaders of PT/EQA programs and providers of control or reference materials, and other related documents. This review found that in general, professional guidelines recommend the use of appropriate QC materials in all assays and the inclusion of positive controls when available. It was also recognized that these guidelines are often developed with considerations for practicality and concurrent QC material availability. For example, recommendations are often made regarding quality assurance alternatives, including rotating positive controls over a reasonable timeframe and the use of synthetic control materials, in situations when it may not be practical to include all positive controls in each run of tests that detect multiple mutations, or when positive controls for all mutations or alterations to be detected are not available. Examples of professional guidelines on use of QC materials in genetic tests are summarized in Tables 3 and 4.
The group felt that existing professional guidelines provide sound guidance for use of QC materials to assure and monitor the quality of clinical genetic tests. However, data are lacking regarding to what extent these guidelines are adhered to, whether barriers exist to implementation of these guidelines, and what the barriers might be. The group recommended that 1) continuing review is appropriate to ensure these guidelines are up to date as technology evolves, and 2) data collection on laboratory QC practices is necessary and helpful for identifying key issues and developing further guidance on use of QC materials in genetic tests.
Regulatory oversight for providers and users of QC materials
To clarify the regulatory environment for providers and users of QC materials for genetic tests, the QC Material Oversight Workgroup reviewed related FDA, CLIA, and State requirements, with an emphasis on regulatory oversight in the U.S.
FDA oversight for nongovernment and commercial providers of QC materials
QC materials, which may be marketed as assayed and unassayed controls, are subject to 21 CFR Part 820 Quality System Regulation. Under this regulation, assayed QC materials need to have manufacturer-assigned analyte values. The target quantitative or qualitative values are for specific system applications and are stated in the product labeling. Unassayed QC materials have no assigned analyte values and no specified system applications; clinical laboratories are responsible for establishing their specific performance parameters. Unassayed QC materials are currently exempt from FDA premarket review.19
Currently, QC materials for in vitro diagnostic (IVD) devices are regulated under 21 CFR 862.1660 for clinical chemistry devices, 21 CFR 862.3280 for clinical toxicology devices, 21 CFR 864.5425 for coagulation and coagulation factor devices, 21 CFR 864.8625 for hematology analyzers, and 21 CFR 866.2480 for culture media for microorganisms. QC materials for clinical chemistry and clinical toxicology are classified as Class I devices, whereas QC materials for coagulation and hematology are Class II devices. For Class I devices, premarket notification must be submitted to FDA if they meet the limitations in 21 CFR 866.9, which include molecular diagnostics and devices for genetic disorders. QC materials could be submitted to and cleared by FDA, either separately or as a component of an assay system, following guidance provided by FDA.20
Regulatory oversight regarding use of QC materials in genetic tests
Requirements under the Clinical Laboratory Improvement Amendments (CLIA)
CLIA sets forth federal standards for laboratories performing patient testing to ensure the quality of laboratory testing in the U.S.21 Requirements regarding use of controls or control materials in the testing process are provided as general requirements under section 493.1256 and, when applicable, are specified for testing specialties and subspecialties. For genetic testing, current CLIA regulations contain requirements for a clinical cytogenetics specialty but no specific requirements formolecular or biochemical genetic testing. Therefore, laboratories performing molecular and/or biochemical genetic testing are at present subject to the applicable general CLIA requirements for quality systems and laboratory personnel, and in some cases to specific specialty or subspecialty personnel requirements as applicable. The general requirements for control procedures and control materials that are applicable to genetic testing include:
-
Control procedures must monitor the test system over time and detect immediate errors due to test system failure, adverse environmental conditions and operator performance. Control materials, which may be supplied by the test system manufacturer or another source, must be tested in the same manner as patient specimens.
-
Laboratories must establish or verify the criteria for acceptability of all control materials. For in-house-developed tests, which include most genetic tests, laboratories must establish the number, type, concentration, and frequency of performance of control materials. Results of control materials must meet the criteria for acceptability before patient test results can be reported.
-
At least two control materials are required for each molecular amplification procedure at least once each day patient specimens are assayed. This requirement applies to molecular testing for heritable conditions, cancers, and infectious diseases. In addition, if reaction inhibition is a significant source of false negative results in a molecular amplification procedure, a control material capable of detecting the inhibition needs to be included in the test procedure.
-
Other general requirements for control materials are also applicable to genetic testing. For example, at least once each day patient specimens are assayed or examined, a negative and positive control material must be included for each qualitative procedure, and two control materials of different concentrations must be included for each quantitative procedure. For each test system that has an extraction phase, two control materials, including one that is capable of detecting errors in the extraction process, must be included at least once each day of patient testing.
-
Control material testing must be rotated over time among all operators performing the patient testing.
-
For test systems that do not have control materials available, the laboratory is responsible for developing an alternative mechanism to detect immediate errors and monitor test system performance over time.
State requirements
Some State agencies regulate clinical laboratories through licensure of facilities and/or personnel. For example, New York and Washington are the two exempt States under CLIA that have State-specific clinical laboratory oversight programs.22 The New York State Clinical Laboratory Evaluation Program (CLEP) provides regulatory oversight to laboratories performing testing for New York State residents.23 For genetic testing, CLEP requirements include specific QC standards, method validation procedures, and personnel qualifications. For each genetic test submitted for CLEP review and approval, laboratories are required to submit procedure manuals that fully describe control materials used for each assay, both for the method validation procedure and for the routine testing process. Similar requirements exist for molecular oncology testing detecting tumor-specific acquired genetic alterations. Table 5 summarizes New York State requirements and guidance for use of control materials in molecular genetic testing, molecular oncology testing, and cytogenetic testing.
Current research efforts to develop QC materials for genetic tests
Current research efforts in QC material development
To facilitate identification of further research needs, the group reviewed a number of current efforts to develop and provide QC materials for genetic tests. These efforts are summarized into three major areas:
-
Development of stably transformed human cell lines with characterized mutations. An example of this approach is the CDC-funded collaborative effort that generated cell lines for use as positive control materials for public health-significant genetic tests.9 The products of this work are now publicly available in the form of cells and DNA samples through the Coriell Cell Repositories.10
-
Development of synthetic or artificially constructed QC materials. This approach develops noncell line-based QC materials using a variety of methodologies such as PCR,15 oligonucleotide synthesis,16 and recombinant DNA techniques.24 The “synthetic” or “artificially constructed” materials are often intended for use in panel or multiplex assays, such as mutation analysis for CF and a number of other diseases;11
Strengths and weaknesses of reviewed approaches
The major advantage of genomic samples, i.e., samples from residual patient materials and human cell lines, is that these materials resemble patient samples that genetic testing laboratories routinely process; therefore, such materials are presently considered a preferred choice for QC and PT/EQA purposes. However, for multiplex or panel-based testing such as CFTR mutation analysis, including genomic control samples for all mutations to be detected may not be always practical or cost-effective, since it is unlikely for a patient sample or unmodified human cell line to contain the desired multiple mutations. Genetic engineering holds the ability to introduce the mutations needed into cell lines to create QC materials that mimic real patient samples; however this approach is technically more difficult and requires significant resources.
Synthetic control materials have the ability to provide cost-effective controls for multiplex or panel-based assays. These controls have the potential to accommodate the rapidly changing technology platforms and are also relatively easy to prepare in large quantities. On the other hand, since synthetic QC materials often do not resemble real patient specimens, they may not be suitable for monitoring certain steps of the testing process and may have limited use in PT/EQA challenges and test validation.
Determining and monitoring genetic tests in urgent need of QC materials
Defining areas of need for QC materials
The group suggested that genetic tests routinely offered in clinical and public health practice should be considered as an initial step in determining tests in the greatest need for appropriate QC materials. It was recognized that while availability of QC materials is in need of improvement for the most common genetic tests, the need for assurance of reliable testing for less common and newly discovered diseases is equally important. In addition, the group discussed the following aspects in defining areas of need for QC materials:
-
Intralaboratory testing process: Appropriate QC materials should be available for test validation, for monitoring the multiple steps of the analytic process during each run of patient testing, and for lot-testing of new reagent batches.
-
Test development: Appropriate QC materials are needed both by IVD manufacturers for developing commercial test kits and by laboratories for in-house development of genetic tests.
-
Performance evaluation: PT/EQA programs need to have access to a sample pool representative of the clinical situations that laboratories may encounter in patient testing, and need to provide both disease-specific and methodology-based challenges.
Prioritizing needs for QC material development
Participants recommended that a priority list be developed for genetic tests in need of appropriate QC materials. From a standpoint of QC material development, the group felt that a few common conditions such as factor V Leiden and prothrombin G20210A thrombophilia, MTHFR thermolabile variant, HFE-associated hereditary hemochomatosis, and sickle cell anemia, in which common mutations account for most or all of the disease-related alleles, would not be among the most urgent needs since sufficient control materials are already publicly available for these tests. The current QC material needs identified by the group include:
-
Genomic and synthetic control materials for the ACMG-recommended panel of CFTR mutations for preconception and prenatal carrier screening, as well as less common mutations included by many laboratories in their assays based on clinical relevance and/or significance for specific subpopulations.7 For genomic QC materials, while the ACMG-recommended mutations are now available from the Coriell Cell Repositories,27 to enable standardized, or more uniform, sizing for the FMR1 CGG repeats for both PCR and Southern blot assays. Controls for determining methylation status of the CGG expansion are also suggested.
-
Control materials for the ACOG-recommended preconception carrier screening testing for individuals of Ashkenazi Jewish descent.28 In light of the upcoming CAP proficiency survey and ACMG technical guidelines, appropriate QC materials would be needed for mutations associated with Tay-Sachs disease, Canavan disease, familial dysautonomia, mucolipidosis IV, Niemann-Pick disease type A, Fanconi anemia group C, Bloom syndrome, and Gaucher disease.
-
QC materials for diagnostic testing for population or subpopulation screening, such as DNA-based testing for galactosemia, medium-chain acyl-CoA dehydrogenase deficiency, biotinidase deficiency, and other newborn screening conditions.
-
QC materials for pharmacogenetic tests, including testing for drug metabolizing enzyme polymorphisms such as CYP2D6, CYP2C9, CYP2C19, and thiopurine S-methyltransferase (TPMT) variants. Control materials need to be developed to represent allele specificity of the CYP loci in various populations and subpopulations.
-
Control materials for mutations associated with mitochondrial disorders, such as Kearns-Sayre Syndrome, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes, Leber hereditary optic neuropathy, and nonsyndromic hearing loss with or without aminoglycoside sensitivity.
-
QC materials for standardizing trinucleotide repeat sizing for Huntington disease, spinocerebellar ataxia type 1 and type 7, and myotonic dystrophy type 1.
-
Appropriate control materials for sequencing assays should be considered a pressing need, due to the increasing use of this methodology in molecular diagnostics. Sequencing controls would also help with test validation and quality assurance for rare disease testing, since many rare genetic diseases involve private mutations for which sequencing is the preferred detection methodology.
Refining and continuously monitoring the priorities
The group recommended that QC challenges and needs for appropriate QC materials be continuously monitored as the field of genetic testing and molecular diagnostics evolves. For example, as microarray-based genomic screening assays are now being commercially developed, problems with control materials are likely to arise when these tests become available for the clinical market. To effectively monitor the needs of the community, periodical surveys could be conducted among users of QC materials, including testing laboratories, test developers, and IVD manufacturers, in collaborations with professional organizations and PT/EQA programs. The group suggested that the following parameters be collectively monitored for ongoing needs assessment, the demand of the tests in clinical and public health practice; recommendations of professional societies regarding test utilization; the need for testing standardization; and QC material needs captured by materials repositories. In addition to DNA-based human genetic testing, upon which the group focused as the initial step of needs consideration, biochemical genetic testing and related areas of molecular diagnostics, such as molecular hematology and oncology testing, were suggested as areas that should be monitored and assessed for needs of QC materials.
Establish a process to facilitate QC material contribution and access
The group considered it critical to establish a process that will facilitate submission of materials to central repositories for public availability after proper validation. Since such a process will entail participation of material contributors, cell banks, and repositories, a workgroup discussion was held to address and coordinate both perspectives. Key factors identified for this process include:
Utilizing available resources of existing cell banks and repositories
This would be a practical approach at present to make needed QC materials broadly available to the community. Ideally, cell repositories should be enriched with generally accepted and/or recommended QC materials, such as cell lines containing mutations or polymorphisms of interest, that have been sufficiently characterized for each test. Cell banks and repositories should work with contributors to overcome logistical issues associated with the submission process and should provide submission kits for sample packaging and shipping.
Logistical issues related to QC material contribution and distribution
Institutional Review Board (IRB) review, human subjects research, informed consent, and privacy issues were specifically considered during the group deliberation as they were identified as the major logistical concerns associated with the material contribution process.
In particular, the following issues were discussed:
-
Could the process of material submission and distribution be restricted to QC uses for clinical genetic testing only? If so, is IRB review necessary?
-
If sample submission to cell repositories warrants IRB review, should it be considered an activity not involving human subjects or exempt from the human subject protection regulations under 45 CFR Part 46, such as the informed consent requirements?
-
How should guidance be developed to help clinical laboratories and investigators to address their logistical concerns?
It was suggested that, considering the broad uses of QC materials in control procedures, test validation, quality assurance, external quality assessment, and test development, it might be difficult to exclude research elements from some of these activities. For example, using residual patient specimens in control procedures for patient testing or QA practices would generally be considered a nonresearch activity; however, in some cases, development of new tests using residual patient specimens could meet the definition of research and necessitate IRB review.29 There are also practical reasons that cell repositories would not or could not restrict the materials they provide for nonresearch uses only. Such restrictions would be difficult to enforce; in addition, since activities including establishment, expansion, and distribution of stably transformed cell lines are currently undertaken by major repositories, such as the Coriell Cell Repositories with funding support from NIGMS and other research grants, it is required that the materials developed be made available to the research community. Therefore, at least at present, limiting material contribution and distribution to nonresearch QC purposes may not be practical or feasible. The group also felt that guidance should be obtained from the Office of Human Research Protections (OHRP) on this issue, to provide clarifications on QC activities that should not be considered research.
The group then discussed whether the process of enriching cell repositories with publicly available QC materials for genetic testing should be considered an activity not involving human subjects, and thereby the human subject protection requirements under 45 CFR Part 46 would not apply.29 While OHRP considers submission of nonidentifiable human materials collected for purposes other than submission to tissue/cell repositories not involving human subjects, it recommends that the determination on human subject's involvement be made by institutions or IRBs, but not by the investigators.31 Based on these considerations, it would be advisable that material contributors consult with their institutions or IRBs regarding specific policies in place and measures to take.
In light of the complexity of these issues, the group felt strongly that guidance should be developed to help potential material contributors to address their logistical concerns and to assure compliance with federal, State, and local requirements. In particular, model protocols should be developed for typical material contribution approaches. For situations requiring IRB review, guidance should be provided regarding appropriate level of review and applicable exemptions from the 45 CFR 46 regulations, such as the exemption under 45 CFR 46.101(b) (4) for collection or study of only existing data, documents, or pathologic or diagnostic specimens.29 If the exemption is approved following IRB review, a material contributor would be able to submit samples or materials to cell repositories without re-contacting patients to obtain individual informed consent.29 These protocols should also consider information to be submitted on normal and abnormal specimens and provide guidance on measures needed for privacy protection, in order to maximize material utility without violating patient rights.
Develop protocols to facilitate clinical laboratories and investigators to contribute materials with mutations of interest
The group discussed a three-pronged strategy of material contribution and the advantages and disadvantages of each approach. It was agreed that defined protocols should be developed for each approach and provided to potential contributors to facilitate the material contribution process.
Specific issues associated with each approach included:
-
Submission of residual patient specimens to cell repositories. After reviewing the success of the CDC-funded collaborative project to establish positive control cell lines from anonymous residual specimens,9 the group recommended that this experience, including the sample submission timeframe, the submission process, and the IRB protocol, be shared with the genetics community as a model process.
-
Submission of existing cell lines to cell repositories. Utilization of existing immortalized cell lines for QC materials may be the simplest approach scientifically. Logistical issues associated with submission to cell repositories would depend upon the initial study design in the research setting and/or the consent agreement in the clinical laboratory; therefore, a review of the existing informed consent terms is needed to determine whether additional consent or IRB approval is warranted. If so, the informed consent agreement, such as an unsigned copy of the consent form, should be provided to the cell repository in submitting each cell line.
-
Submission of materials through targeted collection or re-collection. As discussed in the section above, informed consent would need to be obtained for research purposes to enable sample submission through this approach. The consent form could be developed with checkbox options, with one option for QC purpose only and another for both research and QC purposes. Patient contact or re-contact would be required for sample submission through this approach, providing an opportunity for discussion about uses of the requested samples. The experience of the Coriell Cell Repositories in establishing its cell collections can be utilized as one model for this approach.
Incentive program
The group recommended that an incentive program be developed to encourage and stimulate contribution of QC materials for public availability. Based on a survey conducted by the Coriell Cell Repositories at the 2004 European Society of Human Genetics annual meeting, 72% of respondents considered public access to materials as a fair and reasonable incentive, 22% would be encouraged by a certificate of appreciation, and 17% considered payment an incentive.32 Participants suggested exploring these approaches further as well as additional incentives, which could include 1) nonmonetary reward from cell banks or repositories, such as a complimentary QC sample for each submitted material; 2) recuperation of laboratory and patient costs, such as phlebotomy and transportation costs incurred by the patient, through a funding mechanism yet to be developed; and 3) peer recognition through publications and professional societies.
Engaging the community in material contribution process
The group recommended that web-based resources be developed by CDC to provide information to the genetics community to facilitate material contribution. Another effective means discussed was distributing a letter among members of professional organizations to enlist participation in material contribution. It was agreed that the letter should be distributed together with the submission protocols and other necessary information, such as guidance on informed consent, to help contributors accomplish the task. Workshops and satellite meetings at major professional meetings were suggested as additional mechanisms for information dissemination.
Define validation processes for QC materials
The group agreed upon the overall principle that both commercially available and laboratory-developed QC materials should be appropriately validated before being used in control and/or calibration procedures for patient testing. Current professional guidelines and regulatory oversight are convergent in that QC materials for patient testing should be adequately validated, but no specific guidance for validating or verifying QC materials for genetic tests are available. In addressing the need to develop recommendations for practical, reliable processes for QC material validation, a two-step approach was adopted by considering QC materials for heritable or germline mutation testing as the initial step, and those needed for the more complex somatic mutation testing and quantitative assays as the next step. For this report, recommendations are primarily directed toward the principle and methods of validation and establishment of traceability for control and reference materials used for DNA-based heritable mutation testing.
“Gold standard” for DNA sequence verification
The current “gold standard” for determination and verification of DNA sequences is fluorescent DNA sequencing. For mutations or polymorphisms present in either heterozygous or homozygous state, this method should be adequate and preferred for verifying the presence or absence of sequence variants. At present, however, the technology is size-limited for the quantification of trinucleotide repeats; DNA sequences involving large repeat sizes, such as high premutations or full mutations of the FMR1 gene, may require an alternative verification method.
The group considered the ACMG recommendations for standards for interpretation of sequence variants33 applicable to verification of sequence variants in QC materials as well. For example, the recommendations that reports of sequencing analysis should “clearly identify the gene(s) analyzed, indicate the presence of a sequence variation, and, if applicable, indicate the location of the sequence variation by nucleotide position, codons affected, and amino acid change(s)” and “include the fraction of exon sequence and intron-exon boundary sequence analyzed,”31 should be adopted as criteria for verifying or confirming sequence variants in control or reference materials, with further guidance on criteria for acceptance or rejection of DNA sequence information. Although not widely applied, such acceptance criteria are available for assessing sequence quality and accuracy.34
Traceability for molecular QC materials
Addressed directly or indirectly in FDA regulations and the International Organization for Standardization (ISO) guidelines, establishment of traceability of QC materials is important because the accuracy of laboratory test results depends highly on the traceability of materials used to calibrate, verify and control these tests to primary standards and methods.20,35 Establishment of traceability of reference materials has been carried out by NIST, by European quality standards organizations, and within the medical diagnostic community.
In discussing traceability, the group adopted the concept of “primary reference measurement procedure,” defined as “a reference measurement procedure having the highest metrological qualities, whose operation can be completely described and understood, for which a complete uncertainty statement can be written down in terms of SI units, and where results are, therefore, accepted without reference to a measurement standard of the quantity being measured.”35 It was agreed that the goal of traceability in molecular reference materials is to define, with a specified minimum uncertainty, the nucleic acid sequence and other characteristics within the intended use of the material. Traceability of a nucleic acid sequence is established when a value of certainty of the sequence is assigned based on results of testing with a primary reference method.
Recognizing that the ability to establish traceability depends on the material under consideration, the group made the following recommendations for QC materials commonly used in genetic testing:
-
Cell lines: Traceability of DNA extracted from a cell line can be established by testing with a primary reference method or, if not available, other suitably accurate methods. Since cell lines carry a possibility that new mutations occur during cell division, establishing traceability to a cell line-generated sequence could be problematic, unless the genome of the cell line can be shown to be highly stable over generations.
-
DNA segments cloned into bacterial artificial chromosomes (BAC), yeast artificial chromosomes (YAC), or other constructs: Traceability of DNA extracted from bacteria or yeast can be established by testing with a primary reference method or, if not available, other suitably accurate methods. Traceability of DNA segments in BAC, YAC, and other constructs can be established by the same testing if the genome of the microorganism can be shown to be highly stable over generations.
-
Amplified or synthetic nucleic acid products: Traceability of amplified or synthetic nucleic acid products can be established by testing an aliquot of an amplification or synthesis batch with a primary reference method or, if not available, other suitably accurate methods. Since amplification reactions carry a probability that base incorporation errors occur in an early cycle thereby generating a substantial quantity of unexpected final product, traceability should be established by retesting each batch of amplified product.
Validation process for genetic testing QC materials
It would be desirable for publicly available QC materials to meet applicable US and international standards to facilitate their international acceptance. Based on agreement that validation methods should allow portability and commutability of the QC materials, the group recommended that two levels of validation should be performed. At the first level, reference testing should be conducted by two methods, if possible, to confirm the utility of the material for these methods. Then further validation should be conducted through interlaboratory evaluation, using current test methods and technology platforms. Professional organizations such as ACMG and AMP, PT/EQA programs such as CAP and EMQN, and genetic information resources such as GeneTests and Eurogentest, were suggested as mechanisms for recruiting laboratories to participate in efforts to validate QC materials for genetic testing.
Develop better coordination of funding sources and opportunities
The group strongly advocated that, in addition to the current efforts supported by CDC, NIH, NIST, industry, and others, continuing funding and resources are needed to support additional projects to develop and provide the QC materials that have been identified as high priorities. Efforts should be made to coordinate activities of federal agencies and help determine the level of funding needed to sustain the process for QC materials development. The NIST certificate program was suggested as a mechanism for provision of quality QC materials. Furthermore, mechanisms should be explored to establish public/private partnerships, to engage broader participation of the IVD industry and QC material manufacturers in the process.
CONCLUSIONS
Improving availability of appropriate QC materials is of critical importance for promoting and assuring the quality of genetic testing, developing performance evaluation and PT/EQA programs, and facilitating new test development. Programmatic needs, as identified in this report, should provide directions in which further efforts are called upon. Furthermore, there is a great need for a structured organization to coordinate activities of stakeholders and identify funding venues.
Need for coordination in national and international scope
To meet the needs of the rapidly expanding capacity of genetic testing in clinical and public health settings, a national or international QC material coordination program should be established to 1) monitor community needs on a continuing basis, 2) improve information exchange among users and providers of control and reference materials, and 3) facilitate material development, submission, collection, validation, and distribution. It was suggested that an expert panel be retained to serve as a resource to the coordination program. Based on these recommendations, a Genetic Testing QC Materials Program has been established by CDC in March 2005, to coordinate a self-sustaining community process to improve the availability of appropriate and verified materials to serve these needs.36
Information Dissemination
Information dissemination is critical for engaging the broader community and ensuring funding resources and support. It is important to fully utilize existing information venues, including 1) web-based resources, 2) targeted mailings to potential contributors, users, developers, and providers of QC materials, 3) emails to solicit participation in material contribution and verification, 4) flyers and newsletters, 5) workshops, exhibits, presentations, and posters at professional meetings, and 6) additional QC materials conferences, to ensure a sustained community process. Furthermore, an interactive website should be available to facilitate information exchange and communication among users, providers, contributors, and developers of QC materials. This website should provide information on material availability and verification information, inform the community of materials needed, increase awareness about material contribution processes, provide helpful references and published protocols regarding QC materials development, contribution, and verification, and serve as a mechanism for communication and discussion. As part of the Genetic Testing QC Materials Program, this interactive website has been established by CDC to serve these purposes.36
References
Williams LO, Cole EC, Lubin IM, Iglesias NI, Jordan RL, Elliott LE . Quality assurance in human molecular genetics testing: status and recommendations. Arch Pathol Lab Med 2003; 127: 1353–1358.
Williams LO, Cole EC . General recommendations for quality assurance programs for laboratory molecular genetic tests. Final Monograph. 1999. http://www.phppo.cdc.gov/dls/pdf/genetics/dyncor.pdf Accessed April 2, 2005.
Association for Molecular Pathology. FDA-approved Molecular Diagnostics Tests. Available at: http://www.ampweb.org/FDAtable/FDAtablenov04.htm Accessed November 2, 2004.
Association for Molecular Pathology statement. Recommendations for in-house development and operation of molecular diagnostic tests. Am J Clin Pathol 1999; 111: 449–463.
American College of Obstetricians and Gynecologists, and American College of Medical Genetics. 2001. Preconception and prenatal carrier screening for cystic fibrosis. Clinical and Laboratory Guidelines. American College of Obstetricians and Gynecologists, Washington, DC.
Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ . Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med 2001; 3: 149–154.
Watson MS, Cutting GR, Desnick RJ, Driscoll DA, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Gene Med 2004; 6: 387–391.
CFTR Mutation Panel. Coriell Cell Repositories. http://ccr.coriell.org/nigms/charmut/cfpanel.html. Accessed March 20, 2005.
Bernacki SH, Stankovic AK, Williams LO, Beck JC, et al. Establishment of stably EBV-transformed cell lines from residual clinical blood samples for use in performance evaluation and quality assurance in molecular genetic testing. J Mol Diagn 2003; 5: 227–230.
Coriell Cell Repositories. 2005. CDC Cell and DNA Repository: Validated Materials Available for Use as Positive Controls for Molecular Genetic Testing. Available at: http://locus.umdnj.edu/cdc/ Accessed March 20, 2005.
Jarvis M, Iyer RK, Williams LO, Noll WW, Thomas K, Telatar M, Grody WW . A novel method for creating artificial mutant samples for performance evaluation and quality control in clinical molecular genetics. J Mol Diagn 2005; 7: 247–251.
Coriell Cell Repositories. 2004. http://www.coriell.org/ccr/ccrsumm.html. Accessed April 2, 2005.
Levin BC, Cheng H, Reeder DJ . A human mitochondrial DNA standard reference material for quality control in forensic identification, medical diagnosis, and mutation detection. Genomics 1999; 55: 135–146.
O'Connell CD, Atha DH, Jakupciak JP, Amos JA, Richie K . Standardization of PCR amplification for fragile X trinucleotide repeat measurements. Clin Genet 2002; 61: 13–20.
Lebo RV, Dunphy G, Quicci M, Galehouse D . Multiple synthesized control mutations optimize test quality. Am J Hum Genet 2004; 73 Supp: 414, Abstract #1429.
Christensen TM, Jama M, Ponek V, Lyon E, et al. Design, development, validation and use of synthetic nucleic acid controls for diagnostic purposes and application to CF testing. J Mol Diagn 2004; 6[ 4]: 435, Abstract TT13.
CRMGEN The CRMGEN project, a European project to develop Certified Reference Materials for molecular genetic (DNA) testing . 2005. http://www.crmgen.org. Accessed April 2, 2005.
Eurogentest. Genetic testing in Europe. 2005. http://www.eurogentest.org/testor/cgi-bin/OTmain.php. Accessed April 2, 2005.
Code of Federal Regulations. 21 CFR Part 820 Quality System Regulation. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820&showFR=1. Accessed April 2, 2005.
Food and Drug Administration. Points to Consider Guidance Document on Assayed and Unassayed Quality Control Material; Draft. 1999. http://www.fda.gov/cdrh/ode/99.html. Accessed April 10, 2005.
Code of Federal Regulations. The Clinical Laboratory Improvement Amendments (CLIA). 42 CFR Part 493. http://www.access.gpo.gov/nara/cfr/waisidx_03/42cfr493_03.html. Accessed April 10, 2005.
Washington State Medical Test Site Rules (Chapter 246-338 WAC). http://www.leg.wa.gov/wac/index.cfm?fuseaction=chapterdigest&chapter=246-338. Accessed April 10, 2005.
New York State Clinical Laboratory Evaluation Program (CLEP) Laboratory Standards. http://www.wadsworth.org/labcert/clep/Survey/standardsmenu.htm. Accessed April 10, 2005.
Maine Molecular Quality Controls, Inc. 2005. http://www.mmqci.com. Accessed April 2, 2005.
Sacred Heart Medical Center. Molecular controls http://www.molecularcontrols.com/index.php?module=base&page=home. Accessed April 2, 2005.
AcroMetrix, Inc. http://www.acrometrix.com/index.php. Accessed April 2, 2005.
National Institute of Standards and Technology. SRM 2399 - Fragile X Human DNA Triplet Repeat Standard 2005. https://srmors.nist.gov/view_detail.cfm?srm=2399. National Institute of Standards and Technology, Gaithursburg, MD. Accessed April 2, 2005.
ACOG Committee on Genetics. ACOG committee opinion. Prenatal and preconceptional carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstetrics Gynecol 2004; 104: 425–428.
Code of Federal Regulations. 45 CFR Part 46 Protection of Human Subjects. http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm. Accessed March 10, 2005.
Office for Human Research Protections, Department of Health and Human Services. Guidance on Research Involving Coded Private Information or Biological Specimens. http://www.hhs.gov/ohrp/humansubjects/guidance/cdebiol.pdf. Accessed April 9, 2004.
Office for Human Research Protections. Issues to Consider in the Research Use of Stored Data or Tissues. http://www.hhs.gov/ohrp/humansubjects/guidance/reposit.htm. Accessed April 9, 2004.
Beck JC . Coriell Cell Repositories and the Sample Submission Process. A presentation at the Third QC Materials for Genetic Testing Working Meeting, Nov. 9, Los Angeles, CA. http://www.phppo.cdc.gov/mlp/qcgeneticconference/proceedings.aspx. Accessed April 9, 2005.
ACMG Laboratory Practice Committee Working Group. ACMG recommendations for standards for interpretation of sequence variations. Gene Med 2000; 2: 302–303.
Laboratory of Phil Green. http://www.phrap.org/. Accessed April 1, 2005.
International Organization for Standardization. In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values assigned to calibrators and control materials. ISO 17511: 2003. International Organization for Standardization, Geneva, Switzerland.
Centers for Disease Control and Prevention. Genetic Testing Quality Control Materials Program. 2005. http://www.phppo.cdc.gov/dls/genetics/qcmaterials/default.aspx. Accessed April 12, 2005.
The Clinical and Laboratory Standards Institute (CLSI, formerly NCCLS). MM1-A Molecular Diagnostic Methods for Genetic Diseases; Approved Guideline. CLSI, Wayne, Pennsylvania.
The Clinical and Laboratory Standards Institute (CLSI, formerly NCCLS. MM3-A Molecular Diagnostic Methods for Infectious Diseases; Approved Guideline. CLSI, Wayne, Pennsylvania.
International Organization for Standardization. Terms and definitions used in connection with reference materials. ISO Guide 30: 1992. International Organization for Standardization, Geneva, Switzerland.
Dequeker E, Ramsden S, Grody WW, Stenzel TT, Barton DE . Quality control in molecular genetic testing. Nature Reviews Genetics 2001; 2: 717–723.
National Institute of Standards and Technology. Standard reference materials (SRM)/Definitions http://ts.nist.gov/ts/htdocs/230/232/ABOUT/Definitions.htm. National Institute of Standards and Technology. Gaithersburg, MD. 2005.
College of American Pathologists. College of American Pathologists Molecular Pathology Checklist. http://www.cap.org. Accessed March 20, 2005.
American College of Medical Genetics American College of Medical Genetics Standards and Guidelines for Clinical Genetic Laboratories. http://www.acmg.net/. Accessed April 2, 2005 American College of Medical Genetics. Bethesda, MD.
The European Molecular Genetics Quality Network. Best Practice Guidelines. http://www.emqn.org/bpguidelines.php. Accessed March 20, 2005.
Clinical Molecular Genetics Society. Best Practice Guidelines: Use of the WAVE System in Diagnostic Service. http://www.cmgs.org/BPG/Guidelines/2002/dhplc.htm. Accessed March 20, 2005.
American College of Medical Genetics. American College of Medical Genetics Technical Standards and Guidelines for CFTR Mutation Testing. http://www.acmg.net/Pages/ACMG_Activities/stds-2002/cf.htm. Accessed March 20, 2005.
American College of Medical Genetics. American College of Medical Genetics Technical Standards and Guidelines for Fragile X. http://www.acmg.net/Pages/ACMG_Activities/stds-2002/fx.htm. Accessed April 2, 2005.
Maddalena A, Richards CS, McGinniss MJ, Brothman A, et al. Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the Standards and Guidelines for Clin Genet Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med 2001; 3: 200–205.
American College of Medical Genetics. American College of Medical Genetics Technical Standards and Guidelines for Huntington Disease. http://www.acmg.net/Pages/ACMG_Activities/stds-2002/HD.htm. Accessed March 18, 2005.
The European Molecular Genetics Quality Network Best Practice Guidelines: Familial Breast Cancer (European Guidelines). http://www.emqn.org/guidelines.php?page=brca_eu. Accessed March 18, 2005.
The European Molecular Genetics Quality Network Best Practice Guidelines: Haemoglobinopathies - Guidelines for Carrier Identification and Prenatal Diagnosis. http://www.emqn.org/guidelines.php?page=hbeu1. Accessed March 18, 2005.
The European Molecular Genetics Quality Network Best Practice Guidelines: Hereditary Motor and Sensory Neuropathies (European Guidelines). 2000. http://www.emqn.org/guidelines.php?page=hmsn. Accessed March 18, 2005.
The European Molecular Genetics Quality Network Best Practice Guidelines: Spinal Muscular Atrophy (UK Guidelines). 2001. http://www.emqn.org/guidelines.php?page=sma. Accessed March 18, 2005.
The European Molecular Genetics Quality Network Best Practice Guidelines: Y-chromosome Microdeletions (European Guidelines). 2004. http://www.emqn.org/guidelines.php?page=azf. Accessed March 18, 2005.
Acknowledgements
The authors gratefully acknowledge the contributions of the Association for Molecular Pathology (AMP). Many AMP members participated in the meetings and contributed to these recommendations. We thank the ACMG Laboratory Quality Assurance Committee for support and input; Dr. Richard W. Jenny (New York State Department of Health, Albany, NY), Dr. Devery Howerton and Rhonda S. Whalen (CDC, Atlanta, GA) for their review and insights; and Kathy LaBeau (Washington State Department of Health) for helpful comments on the Washington State oversight for clinical laboratory testing. We would like to acknowledge Dr. Patricia Charache at the Johns Hopkins Medical Institutions for her thoughtful comments and help with the manuscript. We appreciate the input from the following meeting participants: Linda A. Bradley, PhD (CDC, Atlanta, GA); David E. C. Cole, PhD (University of Toronto, Ontario, Canada); W. Andrew Faucett, MS, CGC (CDC, Atlanta, GA); Joan Gordon (Maine Molecular Quality Controls, Inc, Scarborough, ME); Joanne Mei, PhD (CDC, Atlanta, GA); Walter W. Noll, MD, PhD (Myriad Genetic Laboratories, Salt Lake City, UT); Timothy O'Leary, MD, PhD (Veterans Health Administration, Silver Spring, MD); Yvonne Reid, PhD (American Type Culture Collection, Manassas, VA); Kristy Richie, PhD (NIST, Gaithersburg, MD); Timothy Stenzel, MD, PhD (Vysis/Abbott Molecular, Downers Grove, IL); Patrick Terry (Genetic Alliance, Washington, DC); Vincent Vilker (NIST, Gaithersburg, MD); and Janet Warrington, PhD (Affymetrix, Inc, Santa Clara, CA). We are thankful for Colleen Shaw, Andrea Scott, and Candice L. Gardner for their help in organizing and managing the meetings; and for Sabah M. Quraishi for administrative assistance. Meeting sponsors included: Affymetrix, Inc; Baylor College of Medicine; Genzyme Genetics; Laboratory Corporation of America; Mayo Clinic; ARUP Laboratories, BioRad Laboratories, Inc; Quest Diagnostics; Roche Molecular Systems, Inc; and Vysis, Inc. CRMGEN is funded by the Measurement & Testing Activity of the European Commission's 5th Framework “Growth” Programme (EC-Research Directorate General), Contract G6RD-CT-2001-00581. The mention of or reference to commercial products in this report does not imply endorsement by the U.S. government.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Chen, B., O'Connell, C., Boone, D. et al. Developing a Sustainable Process to Provide Quality Control Materials for Genetic Testing. Genet Med 7, 534–549 (2005). https://doi.org/10.1097/01.GIM.0000183043.94406.81
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000183043.94406.81
Keywords
This article is cited by
-
Development of preimplantation genetic testing for monogenic reference materials using next-generation sequencing
BMC Medical Genomics (2024)
-
Research activity and capability in the European reference network MetabERN
Orphanet Journal of Rare Diseases (2019)
-
Reference standards for next-generation sequencing
Nature Reviews Genetics (2017)
-
Quality assurance practices in Europe: a survey of molecular genetic testing laboratories
European Journal of Human Genetics (2012)
-
Establishing a quality assurance plan for nucleic acid-based diagnostic laboratories: from planning to implementation
Accreditation and Quality Assurance (2010)
