
Abstract
Purpose: To identify the gene causing tarsal/carpal coalition syndrome (TCC).
Methods: Individuals from three kindreds with TCC and normal hearing were used to map TCC and screen for mutations in Noggin (NOG).
Results: Three different missense mutations in NOG were found. Two of these mutations are identical to mutations previously reported to cause proximal symphalangism (SYM1).
Conclusions: TCC is allelic to SYM1, and at least two different mutations in NOG can result in either TCC or SYM1 in different families. This finding suggests that phenotypic differences between these conditions are caused by epistatic modifiers of NOG.
Similar content being viewed by others


Main
The genetic analysis of human malformation syndromes characterized by disturbances of skeletal development provides important insights about the control of bone growth and maintenance. Tarsal/carpal coalition syndrome (TCC; OMIM 186570) is an autosomal dominant disorder characterized by fusion of the carpals, tarsals, and phalanges; short first metacarpals causing brachydactyly; and humeroradial fusion.1,2 At birth, all affected individuals have stiffness of the proximal interphalangeal (PIP) joint of the 5th digit with or without bony synostosis. Over time, the distal end of the proximal phalanx and the proximal end of the middle phalanx fuse. As an individual ages, progressive fusion of the PIP joints of digits 4, 3, and 2 proceeds sequentially (Fig. 1). The distal interphalangeal joints are affected less commonly. Humeroradial fusion is not present in infancy but can be noted shortly thereafter. Abnormalities of the elbow are the most variable feature.

Hands of a young woman with TCC. A: Each digit is short and flexion of the PIP joint of the posterior four digits is limited. B: This is illustrated by her attempt to make a fist. Digits, 2, 3, 4, and 5 cannot flex at the PIP joint. C: Radiograph of hands and wrists demonstrating fusion of the PIP joint of each of the posterior four digits, a short 5th metacarpal, and multiple carpal fusions.
The most substantial disabilities suffered by individuals with TCC are caused by the abnormalities of the ankle and foot. Often, ambulating is painful and difficult, necessitating palliative and/or corrective surgical intervention. Height, facial characteristics, and intelligence are normal. Penetrance of the gene appears to be complete, and the clinical findings are similar among males and females.
We collected phenotypic data and biological materials from members of a family with TCC of the “Fuhrmann type” that had been previously reported by Drawbert et al.2 An updated pedigree revealed that 17 individuals in this family (K1) are affected. Data were also collected from two additional unrelated families (K2 and K3) with TCC.
METHODS
Clinical evaluation
All studies were performed with the approval of the Institutional Review Board of the University of Utah and the General Counsel of the Shriners Hospitals for Children. After obtaining informed consent, each participant was evaluated by history and physical examination and/or review of medical records. Radiographs were performed on all individuals. Individuals in three unrelated families in which TCC was segregating in an autosomal dominant pattern were selected for linkage studies and mutation analysis. Twelve of 13 living affected individuals in family K1 and all 6 living affected individuals in K2 were examined by three of the authors (PA, DS, and MB). Both affected individuals in family K3 were examined by DS. The clinical characteristics of many of the affected members of family K1 have been described previously.2 All affected individuals have normal stature and intelligence. One affected individual has a conductive hearing loss caused by absence of the meatus of the auditory canal of each ear. This abnormality was considered a coincidental malformation unrelated to the TCC phenotype.
Ascertainment of genotype
Fifteen milliliters of blood were obtained from all affected individuals and at least their first-degree relatives. Genomic DNA was prepared from lymphocytes3 and cell lines derived from EBV-transformed lymphocytes4 as described elsewhere. Fluorescent-tagged primers were used to amplify genomic DNA in 1X× buffer (10 mM Tris pH 8.3, 50 mM KCl, 1.5 mM MgCl2) using 25 ng of template genomic DNA, 50 μM dNTPs, 20 pmol of each primer, and 1 unit Taq DNA polymerase in a total reaction volume of 25 μL. Samples were cycled 30 times in a Perkin-Elmer 9600 PCR machine using standard profiles. The annealing temperature was decreased 4°C on the sixth cycle. A total of 20 μL of 50% formamide loading dye was added to each reaction following PCR. Samples were denatured for 3 minutes at 94°C and then electrophoresed through 5% denaturing polyacrylamide gels on an ABI automated sequencer.
Linkage analysis
Two-point analysis was performed using MLINK of the LINKAGE package.5 Disease penetrance was set at 0.95 without a sex difference. Normal and disease allele frequencies were set at 0.9999 and 0.0001, respectively. Genotypes for all individuals typed in the reported families are available upon request.
Mutation analysis of NOG
Human NOG coding sequence is contained in a single exon. A 761-bp product containing the NOG coding region was PCR-amplified using previously published primers,6 and the following internal primers (kindly provided by Dr. M. Mangino) were used for sequencing: F1-CAAAGGGCTAGAGTTCTCCGA, F2-CAGCTGCTTCAGTAAGCGCTC, F3-CACCCAGCGACAACCTGC, R1-CCATGAAGCCTGGGTCGTAGT, and R3-CTAGCCCTTTGATCTCGCTCG. Genomic DNA sequences were amplified as described above except for the use of Expand Taq (Boehringer-Mannheim). PCR products were purified on a 2% NuSieve gel and sequenced using ABI Dye-primer or rhodamine sequencing reagents. Sequenced products were loaded on an ABI 377 automated sequencer and analyzed using SEQUENCHER software (Genecodes). The forward and reverse strands of the exon, including flanking splice-recognition sequences, were PCR-amplified and sequenced twice in each direction. The presence of each mutation was confirmed by restriction digest (Fig. 2), and genomic DNA samples from 100 individuals representing 200 control chromosomes were screened for each mutation.

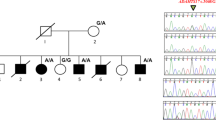
Partial pedigrees, electropherograms, and restriction digests demonstrating mutations in NOG in families with tarsal/carpal coalition with symphalangism syndrome (TCC). Blackened symbols represent affected individuals. Electropherogram of control sequence is in upper panel and affected individual in lower panel. Missense mutation is depicted by arrow in each electropherogram. (K1) A G→T mutation results in a R204L substitution. This mutation creates a new Aci I site that is present in affected individuals (211 bp and 137 bp) but absent in the unaffected individuals (211 bp). (K2) A C→G mutation results in a P35R substitution. Unaffected individuals are homozygous for a Bmy I site (115 bp), whereas loss of this site in affected individuals results in heterozygosity for the 115-bp product and new fragment of 150 bp. (K3) An A→G mutation results in a Y222C substitution. This mutation creates a new Rsa I site that is present in affected individuals (211 bp and 122 bp) but absent in the unaffected individual (211 bp).
RESULTS
Linkage analysis
Initially, we hypothesized that TCC was caused by perturbations of one of the HOX genes. However, before this hypothesis could be tested, Krakow et al.7 reported that a locus for multiple synostosis syndrome (SYNS1; OMIM 186500) mapped to chromosome 17q21, near the locus for proximal symphalangism8 (SYM1; OMIM 185800). Because of the phenotypic similarity of SYNS1 to TCC, we tested markers on 17q for linkage to TCC. A pairwise LOD score of 4.16 was obtained with W359 (AFM268zg9) at a recombination fraction of 0.001. Additional markers spanning ∼40 cM around W359 were tested for linkage and haplotypes constructed on the basis of the marker order estimates of the regional linkage map of chromosome 17. Analysis of recombinants narrowed the critical interval containing the TCC locus between D17S790 and D17S794 (data not shown). This region overlapped the critical intervals of SYM18 and SYNS1,7 suggesting further that TCC might be caused by mutations in NOG.
It is worthwhile to note that at least three other conditions manifest by skeletal abnormalities (i.e., sclerosteosis, van Buchem disease, and familial patella aplasia-hypoplasia) are caused by genes on chromosome 17q21.9–11 This finding suggests that there may be a cluster of genes in this region that participate in skeletal development.
Mutation analysis
Mutation analysis was conducted on at least two affected individuals in each multiplex family. Missense mutations were found in the affected individuals of all three families (Fig. 2). Individuals in family K1 had a G→T mutation that results in a R204L substitution. This mutation has not been reported previously.
Individuals in family K2 had a C→G mutation the results in a P35R substitution. This mutation was reported previously in a sporadic case of SYM1 (family 7 in ref. 6) in which neither parent carried the mutation and paternity was confirmed. Thus, the same mutation appears to have arisen independently in these two families and caused different synostosis syndromes.
Individuals in family K3 had an A→G mutation that results in a Y222C substitution. This mutation was reported previously in a large SYM1 kindred (family 1 in ref. 6) that was used to map the locus for SYM1 by Polymeropoulpos et al.8 Restriction analysis confirmed that each mutation segregated only with affected individuals (Fig. 2). None of the changes that are predicted to cause missense mutations were found in 200 control chromosomes.
DISCUSSION
TCC is one of a heterogeneous group of disorders characterized by multiple synostoses that includes symphalangism-brachydactyly syndrome, WL syndrome,12 deafness-symphalangism syndrome of Herrmann,13 and facioaudiosymphalangism syndrome.14 The pattern of joint involvement was very similar in the families that were studied to define these conditions. Indeed, only the presence of short stature, hearing loss, humeroradial fusion, and brachydactyly varied among them. All of these findings are characteristics of TCC, SYM1, or SYNS1.2,7,8
In contrast to SYM1 and SYNS1, conductive hearing is typically normal in individuals with TCC and fusion of the MP joints is uncommon. SYNS1 is differentiated from SYM1 by consistent involvement of the hips and cervical spine.6 SYM1 and SYNS1 are caused by different missense mutations in NOG,6 suggesting that perturbation of specific domains of the protein that it encodes, Noggin, could underlie the phenotypic differences between these conditions. Furthermore, the observation that only a subset of joints are affected in individuals with SYM1 and SYNS1 also suggested that different tissues might have varied dosage requirements for Noggin and/or that BMP activity is modulated by redundant mechanisms in some tissues. Thus, we hypothesized that TCC is caused by mutations in NOG that perturb Noggin's function in joint formation in the limbs but retain Noggin's role in forming joints of the middle ear ossicles and the vertebral column. This might highlight functionally specific domains of Noggin. We discovered that identical mutations in NOG cause either SYM1 or TCC. However, the phenotypic characteristics of each condition are consistent within a family. This finding suggests that epistatic modifiers are responsible for the varied expressivity and pleiotropy among TCC and SYM1. This is analogous to the finding that, in Nog −/− mice, the severity of the neurological phenotype was dependent on the genetic background of the affected offspring.15
The mechanism by which mutations in NOG cause disease is unknown. Noggin is a member of the TGF-β family of receptors that negatively modulates the activity of bone morphogenetic proteins (BMPs).16 In murine null mutants of Nog17 (i.e., Nog −/− mice), increased BMP activity results in excess cartilage and failure to initiate joint formation, although the spatial expression patterns of BMPs are normal. Because individuals with symphalangism and deletions of chromosome 17q21–22 have been reported,18 it has been suggested that functional haploinsufficiency of Noggin6 disturbs joint morphogenesis and causes multiple synostoses. Alternatively, missense mutations could impair the ability of Noggin protein to participate in dimer formation or in binding to members of the TGF-β superfamily (e.g., BMP2, 4, and 7 in the limb) with which it normally interacts.
In the developing chick and mouse limb, Noggin is expressed in the prechondrogenic condensations and in chondrocytes.19 Targeted disruption of Nog leads to impaired chondrogenesis but does not appear to disrupt patterning of the limb. This finding is consistent with the observation that missense mutations in NOG result in the formation of excess cartilage and progressive bony synostoses, while the patterning of joints in the limb is normal.
A recently proposed model of digit formation20 may explain the earlier onset of fusion in the posterior elements of the hands and feet. In this model, the number and identity of digits is dependent on the activity of Sonic Hedgehog (Shh) and BMP2 in the anterior mesenchyme. Shh acts as a long-range signal to make the anterior mesenchyme competent to respond to a signal from BMP2 and subsequently to induce the expression of BMP2. BMP2 acts in a dose-dependent fashion on the competent mesenchyme to specify digit identity. Lower levels of BMP2 expression specify an anterior identity while higher levels of BMP2 expression promote formation of posterior digits.
The observation that, in individuals with TCC, SYM1, or SYNS1, the most posterior digits are affected at birth is consistent with a higher level of BMP2 activity in the posterior anterior mesenchyme of the developing autopod. A gradient of BMP2 activity could be produced by either higher BMP2 expression or attenuation of BMP2 signaling by inhibitory molecules such as Noggin. However, digit identity is unchanged in individuals with missense mutations in NOG. This finding suggests that establishment of the BMP2 gradient is independent of the function of Noggin and/or other molecules (e.g., Gremlin) can compensate for haploinsufficiency of Noggin.
References
Gregersen HN, Petersen GB . Congenital malformation of the feet with low body height: a new syndrome, caused by an autosomal dominant gene. Clin Genet 1977; 12: 255–262.
Drawbert JP, Stevens DB, Cadle RG, Hall BD . Tarsal and carpal coalition and symphalangism of the Fuhrmann type. J Bone Joint Surg Am 1985; 67: 46–51.
Bell GI, Karem JH, Rutter JR . Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc Natl Acad Sci USA 1981; 78: 5759–5763.
Anderson MA, Gusella JF . Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro 1984; 20: 856–858.
Lathrop GM, Lalouel JM, Julier C, Ott J . Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R, Hudgins L, Cremers CW, Cremers FP, Brunner HG, Reinker K, Rimoin DL, Cohn DH, Goodman FR, Reardon W, Patton M, Francomano CA, Warman ML . Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 1999; 21: 302–304.
Krakow D, Reinker K, Powell B, Cantor R, Priore MA, Garber A, Lachman RS, Rimoin DL, Cohn DH . Localization of a multiple synostoses-syndrome disease gene to chromosome 17q21–22. Am J Hum Genet 1998; 63: 120–124.
Polymeropoulos MH, Poush J, Rubenstein JR, Francomano CA . Localization of the gene (SYM1) for proximal symphalangism to human chromosome 17q21–q22. Genomics 1995; 27: 225–229.
Brunkow ME, Gardner JC, Ness JV, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J . Bone dysplasia sclerosteosis results from loss of the SOST gene product a novel cystine knot-containing protein. Am J Hum Genet 2001; 68: 577–589.
Mangino M, Sanchez O, Torrente I, De Luca A, Capon F, Novelli G, Dallapiccola B . Localization of a gene for familial patella aplasia-hypoplasia (PTLAH) to chromosome 17q21–22. Am J Hum Genet 1999; 65: 441–447.
Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, Hildering P, Vanhoenacker F, Van Camp G, Willems PJ . Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12–q21. Am J Hum Genet 1998; 62: 391–399.
Higashi K, Inoue S . Conductive deafness, symphalangism, and facial abnormalities: the WL syndrome in a Japanese family. Am J Med Genet 1983; 16: 105–109.
Herrmann J . Symphalangism and brachydactyly syndrome: report of the WL symphalangism-brachydactyly syndrome: review of literature and classification. Birth Defects Orig Artic Ser 1974; X: 23–53.
Hurvitz SA, Goodman RM, Hertz M, Bat-Miriam Katznelson M, Sack Y . The facio-audio-symphalangism syndrome: report of a case and review of the literature. Clin Genet 1985; 28: 61–68.
McMahon JA, Takada S, Zimmerman LB, Fan CM, Harland RM, McMahon AP . Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev 1998; 15: 1438–1452.
Zimmerman LB, De Jesus-Escobar JM, Harland RM . The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell 1996; 86: 599–606.
Brunet LJ, McMahon JA, McMahon AP, Harland RM . Noggin, cartilage morphogenesis and joint formation in the mammalian skeleton. Science 1998; 280: 1455–1457.
Park JP, Moeschler JB, Berg SZ, Bauer RM, Wurster-Hill DH . A unique de novo interstitial deletion del(17)(q21.3q23) in a phenotypically abnormal infant. Clin Genet 1992; 41: 54–56.
Pizette S, Niswander L . BMPs are required at two steps of limb chondrogenesis: formation of prechondrogenic condensations and their differentiation into chondrocytes. Dev Biol 2000; 219: 237–249.
Drossopoulou G, Lewis KE, Sanz-Ezquerro JJ, Nikbakht N, McMahon AP, Hofmann C, Tickle C . A model for anteroposterior patterning of the vertebrate limb based on sequential long- and short-range Shh signalling and Bmp signalling. Development 2000; 127: 1337–1348.
Acknowledgements
We thank the families for their participation, generosity, and patience. We thank L.B. Jorde for comments on the manuscript and B. Kramer and W.S. Watkins for technical assistance. This project was supported by Shriners Hospitals for Children (SHC 9510, MB), Primary Children's Foundation (MB), and General Clinical Research Center at the University of Utah (PHS MO1-00064).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Dixon, M., Armstrong, P., Stevens, D. et al. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet Med 3, 349–353 (2001). https://doi.org/10.1097/00125817-200109000-00004
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200109000-00004
Keywords
This article is cited by
-
The earliest known Italian case of bilateral non-osseous calcaneonavicular coalition from the mediaeval cemetery of Troina (Enna, Sicily)
Archaeological and Anthropological Sciences (2021)
-
Novel NOG (p.P42S) mutation causes proximal symphalangism in a four-generation Chinese family
BMC Medical Genetics (2019)
-
TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease
Bone Research (2016)
-
A novel nonsense mutation in the NOG gene causes familial NOG-related symphalangism spectrum disorder
Human Genome Variation (2016)
-
Identification of potential biomarkers for xylene exposure by microarray analyses of gene expression and methylation
Molecular & Cellular Toxicology (2016)
