
Abstract
Purpose:
We investigated whether Brugada syndrome (BrS)-associated variants identified in the general population have an effect on J-point elevation as well as whether carriers of BrS variants were more prone to experience syncope and malignant ventricular arrhythmia and had increased mortality compared with noncarriers.
Methods:
All BrS-associated variants were identified using the Human Gene Mutation Database (HGMD). Individuals were randomly selected from a general population study using whole-exome sequencing data (n = 870) and genotype array data (n = 6,161) and screened for BrS-associated variants. Electrocardiograms (ECG) were analyzed electronically, and data on syncope, ventricular arrhythmias, and mortality were obtained from administrative health-care registries.
Results:
In HGMD, 382 BrS-associated genetic variants were identified. Of these, 28 variants were identified in the study cohort. None of the carriers presented with type 1 BrS ECG pattern. Mean J-point elevation in V1 and V2 were within normal guideline limits for carriers and noncarriers. There was no difference in syncope susceptibility (carriers 8/624; noncarriers 98/5,562; P = 0.51), ventricular arrhythmia (carriers 4/620; noncarriers 9/5,524; P = 0.24), or overall mortality (hazard ratio 0.93, 95% CI 0.63–1.4).
Conclusions:
Our data indicate that a significant number of BrS-associated variants are not the monogenic cause of BrS.
Genet Med advance online publication 06 October 2016
Similar content being viewed by others
Introduction
In recent years, advances in genetics have added new perspectives to our understanding in the field of inherited cardiac diseases associated with sudden cardiac death. The use of genetic information has progressed in the clinical setting and is now broadly used in diagnostics, risk stratification, and therapeutic strategies. Next-generation sequencing has provided a fast and cost-effective approach for large-scale genetic screening. However, its use in diagnostics remains a challenge due to large amounts of genetic information that have to be interpreted to evaluate their pathological relevance.
Recent studies using next-generation sequencing have shown that variants previously assumed to be pathogenic in the setting of Brugada syndrome (BrS) and long QT syndrome are exceedingly overrepresented in the NHLBI GO Exome Sequencing Project population, suggesting that they cannot all be truly causative.1,2,3,4,5
A likely disease-causing mutation is found in approximately 20–30% of probands with BrS, with the majority of these occurring in the SCN5A gene; it is less commonly found in multiple other genes encoding proteins involved in sodium, potassium, and calcium currents. The low yield of genetic testing in BrS is thought to be due to a multitude of undiscovered mutations as well as oligogenic inheritance patterns, where many different genetic risk variants (with different effect sizes on disease susceptibility) are thought to contribute to the disease.6 In the most recent guidelines presented by Priori et al., the diagnosis of BrS is solely based on electrocardiographic findings. However, the results from genetic screening may still have value when the experienced cardiologist has established a substantial clinical index of disease suspicion and as part of family screening of family members of genopositive probands.7 It is therefore important for the genetic information of presumed monogenic inherited variants to be supported by solid evidence to correctly guide risk stratification. Counseling based on potentially false-positive variants in the risk stratification process may lead to misdiagnosis, which can have perilous clinical as well as economic and psychological consequences, not only for the patient but also for the entire family.
In this study, we analyzed age- and gender-stratified randomly selected individuals from the general population, with available phenotypic data and almost complete longitudinal follow-up in registries. The aim was to investigate whether (i) genetic variants previously associated with BrS could be identified in the general population; (ii) carriers of genetic variants previously implicated with BrS have an effect on J-point elevation as well as PR interval, QRS duration, and QTc interval; (iii) carriers of genetic variants previously associated with BrS are more prone to syncope, ventricular tachycardia (VT), and/or ventricular fibrillation (VF) compared with noncarriers; and (iv) carriers of genetic variants previously implicated with BrS have higher overall mortality compared with noncarriers.
Materials and Methods
Identification of BrS-associated variants
All genetic variants previously associated with BrS were identified through a query in the Human Gene Mutation Database (HGMD).8 Only variants labeled “Brugada syndrome” were included. Original articles were systematically reviewed. Familial co-segregation was defined as two or more genotype-positive family members with the phenotype. Positive functional data were defined as any in vitro or in vivo experiment with the minor allele, which differed significantly from the wild-type.
Danish study population
The study cohort was drawn from the Inter99 study.9 The Inter99 study is a randomized, population-based intervention study (CT00289237, ClinicalTrials.gov) including almost 60,000 individuals randomly selected from an age-stratified and sex-stratified sample of the population that investigated the effects of lifestyle intervention on cardiovascular death. The study population comprised individuals in specific age groups (30–60 years) from a defined area of Copenhagen. Genotyping was performed for approximately 10% (n = 6,186) of the individuals. Participants with available DNA samples underwent exome array genotyping (“Exome Chip Population”); a subset of these also had whole-exome sequencing performed (“Exome Sequencing Population”). All participants were of Danish origin by self-report. Figure 1 shows the flowchart of the current study.

Flowchart of the study.
Exome-chip study population
From the Inter99 study, 6,161 individuals were genotyped using the Illumina Human Exome BeadChip v1.0 genotyping array (“the exome chip”), which encompasses approximately 250,000 predominantly rare exonic genetic variants (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). Genotypes were called using GenCall by applying a custom-made cluster file based on 6,000 samples with high-quality data. Quality-control measures included exclusion of samples showing relatedness (first- and second-degree relatives), an extreme inbreeding coefficient (F < 0.1 or F > 0.1), a low call rate (<98%), or a mismatch between sex status in phenotype and genotype data. All examined variants that did not meet Hardy-Weinberg equilibrium were removed (P < 0.01). For variants available on the exome chip, the genotype concordance between the exome sequenced data and the exome-chip data was 0.999. The nonconcordant variants were removed. European ancestry was confirmed using principal-component analysis (Supplementary Figure S1 online).
Whole-exome-sequencing population
From the Inter99 study, 870 metabolically healthy individuals were identified on the basis of normal values of fasting plasma glucose levels, body mass index, and blood pressure. Upon inclusion, these individuals underwent ECG examination and whole-exome sequencing.9,10 The exomes were captured using the Agilent SureSelect All Exon Kit v.2 (46 Mb). Sequencing was performed on an Illumina HiSeq 2000. The mean coverage depth across 46 Mb of capture sequence was 56-fold (56×); average exome coverage per sample was 97.3%. No sample had less than 90% of its exome covered.
European ancestry was confirmed using principal-component analysis. Detailed descriptions of the sequencing procedure as well as quality metrics of the resulting data have been reported previously.10
Electrocardiogram data analysis
Upon ECG analysis, the exome sequenced cohort and the cohort with exome-chip data were pooled. All ECGs were obtained on inclusion as part of the Inter99 protocol and digitally recorded and stored in the MUSE Cardiology Information System (GE Healthcare, Wauwatosa, WI) and later processed using version 21 of the Marquette 12SL algorithm.
ECGs were analyzed electronically. In addition, two experienced physicians analyzed all ECGs manually and independently for the diagnostic type 1 Brugada pattern, the nondiagnostic type 2 Brugada pattern, and incomplete right bundle branch block (IRBBB).
According to current guidelines, type 1 Brugada pattern is characterized by a unique “coved-type” J-point elevation of ≥2 mm and negative T-wave in at least one of the right precordial leads (V1 and/or V2) positioned in the second, third, or fourth intercostal space.11 Type 2 BrS pattern is characterized by J-point and ST segment elevation and a positive T wave in the right precordial leads, with a distinctive “saddleback” pattern.10 Although nondiagnostic, it may warrant further investigation.13 IRBBB was defined as the RSRʹ pattern in V1 or V2 with a QRS duration of <120 ms in the absence of type 1 ECG pattern. This is important because the RSRʹ pattern can be present on baseline 12-lead ECG of patients with BrS.12
Other ECG measurements were assessed digitally, including heart rate, mean PR interval, mean QRS duration, and mean QTc interval. QT interval was corrected for heart rate using Bazett’s formula (QTcB). Sinus bradycardia was defined as a heart rate <60 beats/min.
Syncope, ventricular arrhythmia, and mortality data analysis
Registry data were used to assess whether subjects harboring a BrS-associated variant at the time of inclusion or during follow-up more frequently had a diagnosis of syncope, ventricular arrhythmias, or BrS. In addition, we assessed whether carriers had an increased risk of death from all causes during follow-up compared with noncarriers.
All Danish citizens are assigned a unique and personal civil registration number that can be linked to national registries on an individual level. The Danish National Patient Registry (NPR) contains information on all inpatient and outpatient activities in Denmark since 1978. The civil registration number was used to access registry data in Danish NPR using the International Classification of Diseases, 10th revision (ICD-10).14 We identified all patients who had previously been discharged with or who during follow-up had one of the following ICD-10 codes introduced in 1994: syncope (R559), VT (DI472), VF (DI490B), cardiac arrest (DI469), and BrS diagnosis (DI472M). Only patients who were admitted to a hospital or were seen in an emergency department were given the diagnosis. Medical histories of carriers were assessed and compared with those of noncarriers.
Information regarding death during follow-up was extracted from the Danish Cause of Death Registry.15 The Cause of Death Registry contains data regarding all deaths among Danish residents since 1970. Patients were enrolled from 1999 to 2001 and followed until 17 December 2012.
Statistics
Categorized nominal data were compared using the chi-square test or Fisher’s exact test, when appropriate. Continuous variables were assessed using the t-test.
All ECGs were analyzed using a one-way analysis of variance. ECG measurements were analyzed for each individual variant and as carriers versus noncarriers. Because means are sensitive to extreme values, we also compared the burden of outliers in J-point elevation in lead V1 and lead V2 in carriers and noncarriers by implicating the diagnostic criteria mentioned. Because we mainly report lack of associations, we consider it a more conservative approach not to present P values corrected for multiple testing. Hence, a Bonferroni correction has not been conducted.
Medical history regarding syncope, VT/VF, and Brugada syndrome diagnosis was assessed from 1994 and until the end of follow-up.
For mortality comparison, the Cox proportional hazards regression was used to calculate the hazard ratio and 95% confidence intervals (CI) between BrS variant carriers and noncarriers. Survival data were adjusted using left truncation and right censoring. Follow-up time began on the day of inclusion and ended at death, emigration, or end of follow-up (17 December 2012), whichever came first. The survival plot was conducted to illustrate the probability of survival when harboring a BrS variant compared to not harboring a BrS variant in different age groups.
The two-tailed log-rank test was used to test the difference between the two groups in the survival analysis. A two-sided P value of <0.05 was considered statistically significant. All genetic analyses were performed using PLINK.16 Statistical analyses were conducted using SAS, version 9.4 (SAS Institute, Cary, NC).
Results
Variants identified in Danish exomes and exome-chip data
From the HGMD survey, we identified 382 genetic variants (including missense, nonsense, indels, and splice error mutations) previously associated with BrS. Among the 870 patients who had whole-exome sequencing performed, we identified 23/382 (6.0 %) HGMD-derived genetic variants previously associated with BrS. These 23 variants affected 145 heterozygote carriers, corresponding to a “BrS genotype prevalence” of 1:6 (145/870; 16.7%) (Supplementary Table S1 online).
Among patients with available exome-chip data, we identified 21/382 (5.5%) variants previously associated with BrS. These variants were identified in 804 heterozygote carriers and were screened in 6,161 individuals, corresponding to a genotype frequency of 1:8 (804/6,161; 13.0%). Sixteen of the 21 variants identified in the population with available exome-chip data were also identified in the exome-sequenced data. In other words, five additional variants were identified exclusively on the exome chip. In total, 28/382 (7.3%) variants previously associated with BrS were identified as affecting 624 individuals. More than 50% (15/28; 53.6%) of the variants were present in eight or more individuals (Supplementary Table S1 online). Of these, 26 were missense mutations, one was a nonsense mutation, and one a splice error mutation (Table 2). None of the 624 genopositive individuals had been given the BrS diagnosis in the Danish NPR previously or during follow-up.
The BrS-associated variants were also examined in the Exome Aggregation Consortium (ExAC) database.17 We identified 132/382 (34.5%), of which 52/132 (39%) were identified in frequencies above the phenotype prevalence in the general population (Supplementary Table S2 online). Twenty-seven of the 28 variants identified in the Danish study cohort were also identified in ExAC with transferable minor allele frequencies for the majority (20/28; 71%) of the identified variants (Supplementary Table S1 online).
J-point elevation in Brugada variant carriers
Electrocardiogram data were available electronically for 6,186 individuals. Age, gender ratio, mean heart rate, mean J-point elevation in lead V1 and lead V2, number of individuals with sinus bradycardia, right bundle branch block, and IRBBB stratified by carrier status are presented in Table 1 . J-point elevation stratified by carrier status is presented in Table 2 . Data on PR interval, QRS duration, and QTc interval are summarized in Supplementary Table S3 online.
The mean J-point elevation for all 28 genetic variants previously associated with BrS was normal according to standard guidelines (<2 mm in V1 and/or V2) when comparing mean J-point elevation of BrS carriers with that of noncarriers on a variant-to-variant level ( Table 2 ). Three variants were associated with mean J-points in carriers differing from those of the noncarriers. The variant R1268Q in SCN10A ( Table 2 ), which was identified in 58/6,186 (0.9%) individuals, had a relatively depressing effect on the mean J-point in lead V2 (0.219 vs. 0.396 mm (P = 0.01)) in carriers compared with noncarriers. The other two affecting the J-point, G873S in KCNH2 and K225N in CACNB2 ( Table 2 ), were single-variant findings identified in one and two individuals, respectively.
Among carriers, 1/624 (0.2%) had a J-point in lead V1 above standard guideline limits and 2/624 (0.3%) had a J-point in V2 above standard guideline limits. For noncarriers, the corresponding numbers were 7/5,562 (0.1%) in lead V1 and 48/5,562 (0.9%) in lead V2. There was no significant difference between carriers and noncarriers with regard to the fraction of J-point elevations above standard guideline limits (PV1 = 0.64; PV2 = 0.23).
Observing the overall effect on the mean J-point elevations in lead V1 and lead V2 when all variants were pooled as a group, the numbers were almost identical ( Table 1 ). In Figure 2 , we present the overall distribution of J-point measurements in lead V1. There is an extensive overlap between the J-point elevation in carriers (red) and noncarriers (blue), with almost an identical distribution (P = 0.80). It could be argued that the overall lack of signal in J-point measurements between carriers and noncarriers could be concealed by pooling variants from various genes (other than SCN5A) with a previous dubious role in BrS pathogenicity.18 Thus, a separate analysis was conducted in which ECG measurements of carriers of any BrS-associated SCN5A variant was compared with that of noncarriers (Supplementary Table S4 online). With the exception of the RR interval, there was no significant difference in PR interval, QRS duration, J-point elevation, or QTc-interval.

Distribution of the J-point for carriers (pink) and noncarriers (purple).
Two experienced physicians analyzed all 6,186 ECGs manually for type 1 and type 2 Brugada pattern and IRBBB. Nineteen genopositive individuals (19/624 (3.0%)) presented with an RSRʹ pattern. None of the genopositive individuals presented with either the type 1 or the type 2 Brugada pattern on the ECGs. Among genonegative individuals, 233/5,564 (4.2% (P = 0.17)) presented with IRBBB and one individual displayed the type 1 Brugada pattern. No genonegative individual presented with the type 2 Brugada pattern.
The genetic variation in the 19 BrS-associated genes is also presented in the Supplementary Tables S5 and S6 online with corresponding mean J-point elevation in lead V1 and lead V2 and is stratified by carrier status.
Effect on PR interval, QRS duration, and QTc interval in variant carriers
The SCN10A variant I1225T (identified in eight individuals) had an overall prolonging effect on the mean QTc interval compared with the wild-type variant (444.2 vs. 423.5 ms (P = 0.02)) (Supplementary Table S3 online). Conversely, the variant R1869C in SCN10A, identified in 16 individuals, had an overall shortening effect on the QTc interval (411.1 vs. 423.6 ms (P = 0.03)). The other identified variants had no effect on PR interval, QRS duration, or QTc.
Registry data on syncope, ventricular arrhythmia, and overall mortality
Among individuals carrying a previously BrS-associated variant, 8/624 individuals (1.3%) had experienced syncope compared with 98/5,562 individuals (1.8%) among noncarriers (P = 0.51).
With regard to cardiac arrest, VT, or VF, 4/620 (0.6%) carriers and 19/5,524 (0.3%) noncarriers had received the diagnosis (P = 0.24). None of the individuals had been given the BrS diagnosis.
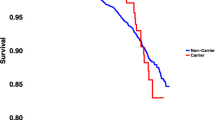
In addition, we compared the overall mortality in carriers and noncarriers ( Figure 3 ). In the carrier group, 28/624 individuals died during a median follow-up time of 13.0 (IQR 12.6–13.4) years, whereas the equivalent all-cause mortality in the noncarrier group corresponded to 259/5,562. The overall mortality was similar between the two groups (hazard ratio = 0.93, 95% CI = 0.6–1.4, P = 0.69).

Survival plot displaying the probability of survival during follow-up in different age groups for carriers (red) and noncarriers (blue) of Brugada syndrome–associated variants. HR, hazard ratio; CI, confidence interval.
Discussion
Using genetic data, we were able to identify 28 genetic variants previously associated with BrS in a study sample consisting of more than 6,000 individuals selected to represent the general Danish population. In this study, we investigated whether individuals harboring a BrS-associated variant had Brugada-associated ECG characteristics. None of the genopositive individuals had the type 1 Brugada pattern. In addition, mean J-point elevation as well as the PR interval, QRS duration, and QTc for carriers compared with noncarriers were similar and normal according to current standard guidelines. Carriers of a BrS-associated variant did not have an increased propensity for syncope or VT/VF, nor did they have increased overall mortality during follow-up compared with noncarriers.
Variants identified in Danish exomes and exome-chip data
The genotype prevalence analyses displayed that one out of six (~17%) individuals in the whole-exome sequenced study sample (n = 870) were carriers of a BrS-associated variant, and one out of eight (~13%) were carriers in the population with exome-chip data (n = 6,161). If the prevalence of BrS is assumed to be 1:2,000,19 then the identified prevalence is markedly higher (300-fold) than expected. In other words, up to 17% of the general population are carriers of a potentially life-threatening mutation. A selective overrepresentation of BrS-associated variants in a cross-sectional Danish study cohort is unlikely. Even when reduced penetrance of certain variants (for some as low as 16%)20 is taken into account, an overrepresentation (60-fold) will still be present.
Nearly all variants identified in the Danish cohort were also found in ExAC with comparable minor allele frequencies, which could indicate that the ExAC database is not enriched with Brugada syndrome and therefore is suitable as a reference for future findings.
The role of variants frequently identified in Brugada syndrome
Because almost half of the variants in this study were rare variants, statistical power in single-variant analysis was limited to variants frequently identified. We have therefore defined a conservative arbitrary cutoff whereby variants identified in eight or more individuals (minor allele frequency ≥0.065%) are set as “variants frequently identified.”
In total, 15/28 variants were frequently identified. Of these, 13/15 (~87%) variants did not have any apparent effect on J-point elevation, PR interval, or QRS duration when inherited in isolation, nor do they predispose to syncope or death. The average age at the end of follow-up among BrS carriers was almost 60 years. In the setting of highly penetrant monogenic causes of BrS, this age is past the typical time of disease onset because BrS patients most often develop symptoms during their fourth decade of life.13
Based on the abundant presence of these more frequent variants identified in a general population (devoid of the BrS diagnosis) with advanced age, innocuous ECG measurements, and no difference in syncope susceptibility, ventricular arrhythmia, and all-cause mortality, it may be argued that these variants are at least not the monogenic cause of BrS.
The role of SCN10A in Brugada syndrome
Hu et al. recently identified SCN10A as a novel susceptibility gene responsible for up to 17% of all BrS cases. 21 In total, 17 variants were implicated with BrS, and the most frequently identified variant in BrS probands was R14L. Functional studies were only conducted using two variants, R14L and R1268Q (the latter only identified in one subject), with a marked reduction in sodium channel current compared with the wild-type model.
In our study, we identified 7/17 of the variants (~40%) identified by Hu et al.21 Six of the variants were identified in 16 or more individuals; R14L was identified in 33 individuals and R1268Q was identified in 58 individuals. None of the individuals presented with a diagnostic BrS ECG pattern. However, R1268Q was associated with a significant depression of the J-point in V2 compared with genonegative individuals ( Table 2 ). In addition, variants I1225T and R1869C were found to have a prolonging and shortening effect on the QTc interval, respectively. All 17 variants have also been identified in the Exome Aggregation Consortium in large numbers (each individual variant in more than 70 individuals) (Supplementary Tables S1 and S2 online).17 In addition, there was no difference in malignant ventricular arrhythmias, nor was there any difference in all-cause mortality in carriers compared with noncarriers, thus questioning the implications of SCN10A in BrS. These findings are in line with recent data published by Behr and colleagues questioning the monogenic role of SCN10A in BrS patients negative for SCN5A mutations.18 Our data indicate that the variants R1268Q, I1225T, and R1869C could represent genetic modifiers of the J-point and QTc interval. The latter two could reflect the fact that BrS-associated genes have overlapping phenotypes (such as long QT syndrome and short QT syndrome) and therefore could represent an entirely different clinical entity.13
The role of low-frequency variants
Interpretation regarding the low-frequency variants is less straightforward. Despite the innocuous ECG findings, these variants can still potentially represent pathogenic variants with incomplete penetrance or genetic modifiers. Many of the low-frequency variants identified in this study have been implicated with BrS through single variants found in BrS cases and the absence of the same variants in a few hundred controls. This highlights the importance of using large ethnically matched control cohorts consisting of thousands of individuals to convincingly be able to associate a genotype with a certain phenotype.
We identified 7.3% (28/382) of all variants previously associated with BrS, which means that the majority (92.7%) of BrS-associated variants were not identified in our study cohort. We are unable to draw any final conclusions for the variants not interrogated in the present study regarding the susceptibility to developing BrS.
Boundaries in genetic screening
Only a small fraction (11/28; ~40%) of all identified genetic variants have been investigated in genetically engineered cell models to elucidate a biologically plausible contribution to pathogenesis (Supplementary Table S1 online).1,3 However, the very same variants failed to induce a significant effect on ECG measurements in this study. A plausible explanation regarding why variants can induce an electrophysiological effect in functional studies but fail to provoke a significant effect on ECG measurements could be that the original studies have not been studied in appropriate model systems engineered to represent human in vivo physiology.
In recent years, many in silico prediction tools have been developed to calculate the functional effects of mutations; however, none of these methods has been tested systematically as valid clinical predictors. This lack of functional and/or biological validation of mutational effects therefore remains one of the most important limitations of genetic test interpretation in cardiac channelopathies. Better variant classification methods are needed to evaluate incidental findings in unselected cohorts before results can be used for patients and participants.
Limitations
Patients, who sought medical attention and were hospitalized were included in the syncope analysis. Individuals who experienced syncope or presyncope and did not seek medical attention or were treated by a general practitioner were not captured; therefore, the incidence of syncope might potentially be underestimated. Using the ICD-10 code for syncope, the diagnosis cannot be differentiated from syncope due to arrhythmia or vasovagal episodes as the primary cause. In addition, registry data regarding syncope were only available from year 1994.
ECGs can be dynamic and can normalize during follow-up; therefore, a type 1 Brugada pattern is only observed in 25% of the tracings. This is a potential limitation because we do not have access to ECGs recorded on different occasions over time.
Because it was not possible to perform a provocative drug test with a class I antiarrhythmic drug in genopositive individuals, the effect may have been underestimated.
In addition to exome sequencing data, we used exome-chip data, for which variants of interest are selected beforehand. Owing to this preselection, variants not represented on the chip are bypassed. Moreover, because most of the genetic variants identified in our study are rare, we are limited by the sample size and even larger data sets are needed to understand the “true” pathogenicity of variants identified in low numbers.
It has previously been reported that BrS is an extremely rare disease in Denmark.22 Thus, it may be argued that a study based on Danish subjects is not representative of other Western countries. We identified one genonegative individual with spontaneous type 1 BrS pattern, corresponding to a prevalence of ~1:6,000. This estimation is in agreement with other approximations of the type 1 BrS pattern in apparently healthy European individuals, indicating that our population is not very different from other European populations in terms of BrS ECG patterns.23,24 Importantly, our findings are based on European individuals, and the results cannot easily be extrapolated to other non-European populations.
Conclusion
We present ECG data and registry data indicating that a significant number of BrS-associated variants had no effect on J-point elevation, syncope propensity, VF/VT susceptibility, or overall mortality. Based on the frequency of individual gene variants, we suggest that a substantial number of variants frequently identified through genetic screening are less likely to associate with a highly penetrant monogenic cause of BrS.
Disclosure
J.H.S. is a member of the advisory board of Medtronic and has received research grants from Medtronic, Biotronik, and Gilead. The other authors declare no conflict of interest.
References
Risgaard B, Jabbari R, Refsgaard L, et al. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 2013;84:489–495.
Refsgaard L, Holst AG, Sadjadieh G, Haunsø S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet 2012;20:905–908.
Ghouse J, Have CT, Weeke P, et al. Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J 2015;36:2523–2529.
Le Scouarnec S, Karakachoff M, Gourraud JB, et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet 2015;24:2757–2763.
NHLBI GO Exome Sequencing Project (ESP). Exome Variant Server. http://evs.gs.washington.edu/EVS/. Accessed 15 May 2015.
Bezzina CR, Barc J, Mizusawa Y, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 2013;45:1044–1049.
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932–1963.
Human Gene Mutation Database (HGMD). http://www.biobase-international.com/product/hgmd. Accessed 1 May 2015.
Glümer C, Jørgensen T, Borch-Johnsen K ; Inter99 study. Prevalences of diabetes and impaired glucose regulation in a Danish population: the Inter99 study. Diabetes Care 2003;26:2335–2340.
Lohmueller KE, Sparsø T, Li Q, et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet 2013;93:1072–1086.
Priori SG, Blomström-Lundqvist C, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2793–2867.
Bayés de Luna A, Brugada J, Baranchuk A, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report. J Electrocardiol 2012;45:433–442.
Hedley PL, Jørgensen P, Schlamowitz S, et al. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat 2009;30:1256–1266.
Lynge E, Sandegaard JL, Rebolj M. The Danish National Patient Register. Scand J Public Health 2011;39(suppl 7):30–33.
Helweg-Larsen K. The Danish Register of Causes of Death. Scand J Public Health 2011;39(suppl 7):26–29.
Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575.
Exome Aggregation Consortium (ExAC). http://exac.broadinstitute.org. Accessed 3 October 2015.
Behr ER, Savio-Galimberti E, Barc J, et al.; UK10K Consortium. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 2015;106:520–529.
Watanabe H, Minamino T. Genetics of Brugada syndrome. J Hum Genet 2016;61:57–60.
Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res 2013;161:1–14.
Hu D, Barajas-Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79.
Holst AG, Jensen HK, Eschen O, et al. Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: a nationwide study. Europace 2012;14:1025–1029.
Gallagher MM, Forleo GB, Behr ER, et al. Prevalence and significance of Brugada-type ECG in 12,012 apparently healthy European subjects. Int J Cardiol 2008;130:44–48.
Letsas KP, Gavrielatos G, Efremidis M, et al. Prevalence of Brugada sign in a Greek tertiary hospital population. Europace 2007;9:1077–1080.
Acknowledgements
This work was supported by the Danish National Research Foundation Centre for Cardiac Arrhythmia, the John and Birthe Mayer Foundation, the Arvid Nilsson Foundation, the A.P. Møller Foundation for the Advancement of Medical Science, the Danielsen Foundation, the Danish Strategic Research Foundation (HEARTSAFE), and Fondsbørsveksler Henry Hansen og Hustru Karla Hansen, født Westergaards Legat. The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (http://www.metabol.ku.dk). The study was partially funded by the Lundbeck Foundation (the Lundbeck Foundation Centre for Applied Medical Genomics in Personalised Disease Prediction, Prevention and Care (LuCamp)).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure and Tables
(DOCX 772 kb)
Rights and permissions
About this article

Cite this article
Ghouse, J., Have, C., Skov, M. et al. Numerous Brugada syndrome–associated genetic variants have no effect on J-point elevation, syncope susceptibility, malignant cardiac arrhythmia, and all-cause mortality. Genet Med 19, 521–528 (2017). https://doi.org/10.1038/gim.2016.151
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.151
Keywords
This article is cited by
-
Reappraisal of variants previously linked with sudden infant death syndrome: results from three population-based cohorts
European Journal of Human Genetics (2019)
-
Paediatric genomics: diagnosing rare disease in children
Nature Reviews Genetics (2018)
-
Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation
Genome Medicine (2017)
