
Abstract
Purpose:
The aim of this study was to characterize the pathogenesis of low bone mineral density in glycogen storage disease type Ia and Ib.
Methods:
A retrospective chart review performed at the University of Florida Glycogen Storage Disease Program included patients with glycogen storage disease type Ia and Ib for whom dual-energy X-ray absorptiometry analysis was performed. A Z-score less than −2 SD was considered low. Analysis for association of bone mineral density with age, gender, presence of complications, mean triglyceride and 25-hydroxyvitamin D concentrations, erythrocyte sedimentation rate, duration of granulocyte colony–stimulating factor therapy, and history of corticosteroid use was performed.
Results:
In glycogen storage disease Ia, 23/42 patients (55%) had low bone mineral density. Low bone mineral density was associated with other disease complications (P = 0.02) and lower mean serum 25-hydroxyvitamin D concentration (P = 0.03). There was a nonsignificant trend toward lower mean triglyceride concentration in the normal bone mineral density group (P = 0.1).
In patients with glycogen storage disease type Ib, 8/12 (66.7%) had low bone mineral density. We did not detect an association with duration of granulocyte colony–stimulating factor therapy (P = 0.68), mean triglyceride level (P = 0.267), erythrocyte sedimentation rate (P = 0.3), or 25-hydroxyvitamin D (P = 0.63) concentration, and there was no evidence that corticosteroid therapy was associated with lower bone mineral density (P = 1).
Conclusion:
In glycogen storage disease type Ia, bone mineral density is associated with other complications and 25-hydroxyvitamin D status. In glycogen storage disease type Ib, bone mineral density was not associated with any covariates analyzed, suggesting multifactorial etiology or reflecting a small sample.
Genet Med 2012:14(8):737–741
Similar content being viewed by others


Introduction
Glycogen storage disease type I (GSD I) is a disorder of glycogenolysis characterized by accumulation of glycogen and fat in the liver and kidneys. Two subtypes of GSD I exist. GSD Ia is caused by a deficiency in the enzyme glucose- 6-phosphatase, which hydrolyzes glucose-6 phosphate (G6P) to produce glucose in the lumen of the endoplasmic reticulum. GSD Ib is caused by deficient activity of the glucose-6-phosphatase transporter that brings G6P from the cytoplasm into the endoplasmic reticulum lumen. Glucose-6-phosphatase and the glucose-6-phosphatase transporter form the complex responsible for glucose production through glycogenolysis and gluconeogenesis. Clinical manifestations of GSD I include severe hypoglycemia, hepatomegaly, hyperlactatemia, hyperuricemia, and hyperlipidemia.1 GSD Ib is distinguished from GSD Ia by myeloid dysfunction, including chronic neutropenia and functional deficiencies of neutrophils and monocytes. As such, patients with GSD 1b often develop recurrent bacterial infections and a Crohn disease–like inflammatory bowel disease (IBD).2,3,4
Although data are limited, bone-related complications appear to be universal clinical manifestations of GSD I. One study found low radial bone mineral content in 10 of 11 subjects using single-photon absorptiometry to measure bone mass in children with GSD I.5 Similarly, a study that included six patients with GSD I found that all had low or very low bone mineral density (BMD) by dual-energy X-ray absorptiometry (DXA). This study defined low BMD as >1 but <2.5 SD below the mean for age- and gender-matched controls, and very low BMD was defined as >2.5 SD below the mean.6 A third study reported significantly lower BMD in adolescents and adults with GSD Ia as compared with healthy controls, but no difference was seen in prepubertal children with the disease as compared with unaffected controls.7 However, the factors responsible for decreased BMD in GSD I have yet to be elucidated. Poor metabolic control has been implicated, but it has not been shown whether aggressive therapy with improved metabolic control is associated with normal BMD.8 Furthermore, BMD has not been characterized in patients with GSD Ib, as few studies have included subjects with this subtype.5,8
Recent progress in nutritional and supportive management of the GSDs has allowed better control of hypoglycemia and metabolic abnormalities. The purpose of this study was to investigate modifiable factors and the role of metabolic control in the pathogenesis of osteoporosis in GSD I.
Materials and Methods
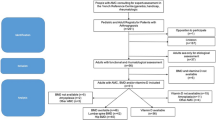
Approval for the study was obtained from the institutional review board at the University of Florida. Informed written consent was obtained from all participants, with assent obtained from minor subjects capable of affirming their desire to participate. A retrospective chart review was performed for 136 patients with GSD Ia and 35 patients with GSD Ib followed at the University of Florida Glycogen Storage Disease Program. The diagnosis of GSD was established either biochemically or by assessment of enzyme testing from a liver biopsy, and was confirmed by mutation analysis. Patients were included if BMD assessment using DXA was performed. DXA scan assessment took place at multiple centers in the United States, and measurements were taken at the L1–L4 vertebrae plus either the left hip or total body less head according to the center’s protocol. The manufacturers’ normative data for the hip in prepubertal children are limited, and evaluation of the hip is not recommended; when measurements were taken at the lumbar spine and hip, only the lumbar spine measurements were included in our analysis. Low BMD was defined as >2 SD below the mean for age- and gender-matched controls using Z-score comparisons. For statistical analysis, all T-scores for adult patients were converted to Z-scores when only T-scores were reported. We corrected for bone age whenever possible. Bone age studies were available for seven of nine children with GSD Ia, and none were delayed as compared with chronologic age. Bone age studies were available for two of four children with GSD Ib, and one was delayed as compared with chronologic age. Data were available for 42 patients with GSD Ia and 12 patients with GSD Ib. For almost all patients, only one DXA scan was available for analysis. If multiple DXA scans were available, the lowest Z-score was included in our study.
Patients were evaluated annually with laboratory testing for parameters indicative of metabolic control, and data were collected from the 5-year period preceding DXA analysis. Metabolic control was determined by mean serum triglyceride (TG) concentration and presence of long-term complications of GSD, including hepatocellular adenoma, microalbuminuria, and proteinuria. Mean serum TG concentration was targeted as a surrogate marker of metabolic control because serum glucose, lactate, and pH fluctuate rapidly, and because serum urate is an unreliable marker in patients treated with allopurinol. A serum TG concentration <500 mg/dl was used to indicate good metabolic control, a cutoff value that was determined at the 2010 Association for Glycogen Storage Disease consensus panel discussion. Mean serum 25-hydroxyvitamin D (25OHD) concentration was determined from annual measurements. In patients with GSD Ib, duration of granulocyte colony–stimulating factor (G-CSF) therapy, mean serum erythrocyte sedimentation rate and history of corticosteroid use for IBD were considered.
Statistics
We analyzed the association between BMD and covariates using a multiple linear regression model. Analysis was performed using R statistical software, manufactured by the R Development Core Team (Vienna, Austria). For GSD Ia, analysis was performed using age, gender, mean serum TG concentration, mean serum 25OHD concentration, and presence of other complications as covariates. For GSD Ib, analysis was performed using age, gender, mean TG concentration, mean 25OHD concentration, duration of G-CSF use, and history of steroid use as covariates. Additional analysis was performed for comparison purposes assessing differences in numerical variables between the low BMD group and the normal BMD group with unpaired two-tailed Student’s t-tests, and categorical variables were assessed using a Fisher’s exact test. We considered a P value <0.05 as statistically significant.
Results
Of 42 patients with GSD Ia (26 male and 16 female) included in the analysis, 23 had low BMD (55%) ( Table 1 ). The median age at BMD assessment was 28.8 years (range 10.5–54 years). Fractures occurred in four individuals with low BMD as a result of minimal or no trauma. Long-term complications were observed more frequently in those with low BMD. In the low BMD group, 12/23 (52.2%) had multiple other complications as compared with 2/19 (10.5%) in the group with normal BMD (P = 0.02). There was a nonsignificant trend toward higher mean serum TG concentration in the group with low BMD (518 mg/dl as compared with 398 mg/dl, P = 0.1). Low BMD was associated with poor metabolic control (defined as mean serum TG concentration >500 mg/dl) and multiple complications in 16/38 individuals (42.1%) for whom data were available. In contrast, only 6/38 (15.8%) had normal bone density in the setting of poor control and multiple other complications (P = 0.018). Mean serum 25OHD level was 19.75 ng/ml in the group with low BMD as compared with 25.75 ng/ml in the group with normal BMD (P = 0.03).
Among 12 patients with GSD Ib (5 male and 7 female), 8 (66.7%) had low BMD ( Table 2 ). The median age at evaluation with DXA scan was 29.7 years (range 10.3–40 years). Fragility fractures were seen in two individuals with low BMD. Using the multiple linear regression model, we did not detect a significant association between BMD and any of the covariates selected for analysis. We did not detect a difference in duration of G-CSF therapy between those with normal BMD and those with low BMD (11.8 years as compared with 14.1 years, respectively, P = 0.68). Mean serum TG concentration was similar among groups. In the group with low BMD, mean serum TG concentration was 281 mg/dl as compared with 169 mg/dl in the normal BMD group (P = 0.267). We did not detect a difference in mean serum 25OHD concentration between the group with low BMD (26.23 ng/ml) and the group with normal BMD (22 ng/ml, P = 0.63). Mean serum erythrocyte sedimentation rate was not significantly different between groups (70 mm/h in the low BMD group and 55 mm/h in the normal BMD group, P = 0.3), and IBD requiring corticosteroid therapy (three in the low BMD group, one in the normal BMD group) was not associated with lower BMD (P = 1).
More than one DXA study was available for eight subjects, including four with GSD Ia and four with GSD Ib. Increased BMD at follow-up evaluation was seen in four of the eight patients, including two with GSD Ia and two with GSD Ib. Treatment with bisphosphonate therapy was initiated in three of the four patients who demonstrated improved BMD over time after the initial DXA scan revealed osteoporosis. The fourth patient, a young woman with GSD Ib, had increased Z-score at her lumbar spine from −2.3 SD at age 24 years to −1.5 SD at age 28 years. Her mean serum TG concentration in the 5-year period preceding her initial DXA scan was 744 mg/dl, as compared with 364 mg/dl in the period between her first and second DXA evaluations. Her vitamin D status and IBD symptoms also improved during the interval between studies. Of the remaining four individuals with serial DXA assessments, three had stable bone density at follow-up. One individual with GSD Ia experienced decreased BMD over a 5-year period, and bisphosphonate therapy was initiated after the second DXA study.
No subjects received bisphosphonate therapy during the period that data were collected for inclusion in this study. However, four adult patients with GSD Ia and one adult patient with GSD Ib were treated with bisphosphonates after DXA analysis revealed osteoporosis. Subsequent DXA scans demonstrated increased bone density in three patients for whom data were available.
Discussion
Low BMD is frequently observed in GSD I. However, the natural history and pathophysiology of osteopenia and osteoporosis in GSD remain poorly understood. This study confirms that low BMD remains a common complication of GSD Ia and Ib; however, we have identified modifiable factors, particularly in GSD Ia, that may be associated with low BMD. Our findings support a link between good metabolic control, defined by primary and secondary surrogate markers, and normal bone density in GSD Ia.
The metabolic derangements that occur with suboptimal control may contribute to bone disease in GSD I through mechanisms of hypoglycemia counterregulation. Glucagon is produced in response to hypoglycemia, which activates cyclic adenosine monophosphate formation in the liver through G-protein-coupled receptors that increase cellular G6P. G6P formed by the mobilization of glycogen cannot be converted to free glucose in GSD I; however, it can be converted to pyruvate, which is reduced to lactate, or it can be irreversibly converted to acetyl coenzyme A by oxidative decarboxylation. Acetyl coenzyme A can shunt into the lipid biosynthesis pathway to generate fatty acids. The lactic acidosis that results from shunting of G6P down the glycolytic pathway causes mineral loss from bone by inducing bone mineral dissolution directly, enhancing osteoclast-mediated bone resorption, and inhibiting osteoblast-mediated bone formation.9 Cortisol is also released in response to chronic hypoglycemia, and glucocorticoid excess has been shown to directly reduce osteoblast production and decrease osteoblast lifespan, whereas the lifespan of osteoclasts is prolonged.10 Therefore, with long-term exposure, the number of osteoclasts is maintained but the number of osteoblasts is decreased and bone formation is substantially reduced.11
Patients with GSD I are prone to low vitamin D concentrations due to the dietary restriction of lactose and galactose, and we report that higher 25OHD concentrations are associated with normal bone density. This common deficiency was recently characterized by Banugaria et al.12 Dietary treatment of GSD I requires restriction of galactose-containing foods, as consumption of this nonutilizable sugar leads to increased G6P formation, increased glycogen formation, and shunting into alternating pathways. Restriction of lactose and galactose can lead to inadequate calcium intake and predisposes individuals to low serum 25OHD concentrations due to insufficient intake of foods fortified with vitamin D. Supplementation of calcium and vitamin D is almost universally required in patients with GSD I to achieve adequate stores if bone density is to be maximized. Notwithstanding the complicated relationship between circulating 25OHD and skeletal health, low serum 25OHD concentrations may reflect suboptimal adherence to medical and dietary treatment, predisposing individuals to low BMD by various mechanisms already described.
Recent progress in nutritional and supportive management of GSDs has allowed better control of hypoglycemia, acidosis, and other metabolic abnormalities. With advances in therapy that make optimal biochemical control of GSD possible, fixing the metabolic abnormalities may decrease the risk of long-term complications including osteoporosis and osteopenia. An association between elevated lactate concentrations and an increased frequency of long-term complications in patients with GSD Ia was noted in 2002.13 Wang et al.14 recently demonstrated that improved metabolic control can decrease the risk of hepatocellular adenoma development in GSD I. Using 5-year mean serum TG concentration as a marker of metabolic control, they demonstrated a significant difference in progression to adenoma formation in the group with mean serum TG concentration >500 mg/dl as compared with those with mean serum TG concentration <500 mg/dl. Similarly, renal function in GSD I has been associated with metabolic control, and optimal metabolic control has been reported to have a protective effect on the development of microalbuminuria and proteinuria in GSD I patients.15 With our study demonstrating a relationship between optimal metabolic control and normal BMD, our data add to the growing body of literature that supports evidence of an association between improved metabolic control and decreased long-term complications.
BMD has not been well characterized in patients with GSD Ib, as few studies have included subjects with this subtype.5,8 This study represents the largest number of subjects with GSD Ib ever reported in the literature. Although we did not find a significant association between bone density and factors related to metabolic control, G-CSF use or severity of IBD in GSD Ib, the trend toward lower mean serum TG concentration, lower mean serum erythrocyte sedimentation rate, shorter duration of G-CSF use and less corticosteroid use in patients with normal BMD may suggest that one or more of these factors are related. Bone-related complications in GSD may be multifactorial, and further studies are needed to clarify the pathophysiology of osteopenia and osteoporosis in these individuals.
The limitations of our study include a small sample size, which can be attributed to the rarity of this disease. In addition, for comparison purposes, we performed a Fisher’s exact test dividing subjects into low and normal bone density groups, and compared them based on the presence of multiple complications and metabolic control together. This allowed comparison with the factors that were deemed to be most important, but there almost certainly are other variables that were not appreciated or compared that likely impact on bone density. Weight-bearing exercise, for example, is one of the most significant factors impacting BMD. Our retrospective review is limited by the inability to include activity level in our analysis. Although we tried to include data for all covariates obtained in the 5-year period before bone density was measured, some data were missing for patients who did not follow up annually as recommended, and this reduced our sample size available for analysis. We routinely obtain DXA scans on all patients with GSD I after growth is complete; however, some DXA evaluations performed at local centers may have been unintentionally excluded from our analysis if results were not transmitted to our center. Furthermore, the fact that we follow patients from diverse geographical locations makes it impossible to control for environmental and other potential factors that may contribute to bone strength.
Conclusion
There is increased evidence that outstanding metabolic control in patients with GSD can reduce or prevent complications. In this study, poor metabolic control was associated with decreased BMD, but near-normal bone density could be demonstrated when vitamin D status and metabolic control were optimized. With the potential risks of pharmacologic therapies in people with liver disease, prevention of osteopenia and osteoporosis is critical. Improved understanding of the pathogenesis of this complication should allow new strategies for improving bone density in this population. Future longitudinal studies and larger sample sizes may allow better understanding of factors that impact bone density, and use of international collaborations may facilitate further investigation.
Disclosure
The authors declare no conflict of interest.
References
Wolfsdorf JI, Weinstein DA . Glycogen storage diseases. Rev Endocr Metab Disord 2003;4:95–102.
Beaudet AL, Anderson DC, Michels VV, Arion WJ, Lange AJ . Neutropenia and impaired neutrophil migration in type IB glycogen storage disease. J Pediatr 1980;97:906–910.
Gitzelmann R, Bosshard NU . Defective neutrophil and monocyte functions in glycogen storage disease type Ib: a literature review. Eur J Pediatr 1993;152 (suppl 1):S33–S38.
Roe TF, Thomas DW, Gilsanz V, Isaacs H Jr, Atkinson JB . Inflammatory bowel disease in glycogen storage disease type Ib. J Pediatr 1986;109:55–59.
Lee PJ, Patel JS, Fewtrell M, Leonard JV, Bishop NJ . Bone mineralisation in type 1 glycogen storage disease. Eur J Pediatr 1995;154:483–487.
Cabrera-Abreu J, Crabtree NJ, Elias E, Fraser W, Cramb R, Alger S . Bone mineral density and markers of bone turnover in patients with glycogen storage disease types I, III and IX. J Inherit Metab Dis 2004;27:1–9.
Rake JP, Visser G, Huismans D, et al. Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: a cross-sectional and longitudinal study. J Inherit Metab Dis 2003;26:371–384.
Schwahn B, Rauch F, Wendel U, Schönau E . Low bone mass in glycogen storage disease type 1 is associated with reduced muscle force and poor metabolic control. J Pediatr 2002;141:350–356.
Krieger NS, Sessler NE, Bushinsky DA . Acidosis inhibits osteoblastic and stimulates osteoclastic activity in vitro. Am J Physiol 1992;262(3 Pt 2):F442–F448.
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC . Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 1998;102:274–282.
Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS . Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology 2006;147:5592–5599.
Banugaria SG, Austin SL, Boney A, Weber TJ, Kishnani PS . Hypovitaminosis D in glycogen storage disease type I. Mol Genet Metab 2010;99:434–437.
Weinstein DA, Wolfsdorf JI . Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr 2002;161 (suppl 1):S35–S39.
Wang DQ, Fiske LM, Carreras CT, Weinstein DA . Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr 2011;159:442–446.
Martens DH, Rake JP, Navis G, Fidler V, van Dael CM, Smit GP . Renal function in glycogen storage disease type I, natural course, and renopreservative effects of ACE inhibition. Clin J Am Soc Nephrol 2009;4:1741–1746.
Acknowledgements
Support for this project was provided in part by National Institutes of Health and National Center for Research Resources Clinical and Translational Science Awards grant 1UL1RR029890. Additional philanthropic support was provided from the Scott Miller Glycogen Storage Disease Program Fund, the Matthew Ehrman Fund for GSD research, and the Jonah Pournazarian Type Ib GSD Fund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Minarich, L., Kirpich, A., Fiske, L. et al. Bone mineral density in glycogen storage disease type Ia and Ib. Genet Med 14, 737–741 (2012). https://doi.org/10.1038/gim.2012.36
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.36
Keywords
This article is cited by
-
Endocrine involvement in hepatic glycogen storage diseases: pathophysiology and implications for care
Reviews in Endocrine and Metabolic Disorders (2024)
-
A female patient with GSD IXc developing multiple and recurrent hepatocellular carcinoma: a case report and literature review
Human Genome Variation (2021)
-
A preliminary study of telemedicine for patients with hepatic glycogen storage disease and their healthcare providers: from bedside to home site monitoring
Journal of Inherited Metabolic Disease (2018)
-
Bone health in patients with inborn errors of metabolism
Reviews in Endocrine and Metabolic Disorders (2018)
-
Long‐term complications of glycogen storage disease type Ia in the canine model treated with gene replacement therapy
Journal of Inherited Metabolic Disease (2018)
