
Abstract
Abstract: Sudden cardiac death is a major contributor to mortality in industrialized nations; in fact, it is the cause of more deaths than acquired immune deficiency syndrome, lung and breast cancer, and stroke together. Frequently, the autopsy becomes the principal diagnostic tool because macroscopic and microscopic analyses reveal the underlying cause of death. However, a significant number of sudden cardiac deaths remain unexplained. These cases are referred to as “natural” or arrhythmogenic. In the young, in up to 50% of sudden cardiac death cases, sudden death is the first and only clinical manifestation of an inherited cardiac disease that had remained undetected by conventional clinical investigations. To improve diagnosis, genetic testing has recently been added to these clinical tools. During the last two decades, there has been considerable progress in the understanding about genetics of sudden cardiac death. With that new information, the probands and their family members can make an informed decision regarding their care and know whether and to what extent they are at risk of suffering from the disease. Thus, genetic technology and expertise have become essential for the diagnosis of some forms of inherited cardiac diseases and to provide a basis for subsequent prevention strategies. This review focuses on recent advances in the understanding of cardiopathies owing to genetic investigations.
Similar content being viewed by others

Main
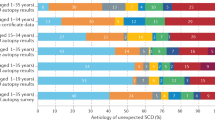
Sudden cardiac death (SCD) is responsible for ∼1 million deaths annually in the developed world,1 which makes arrhythmias as one of the most significant causes of death and disease in the general population. Most cases of SCD are related to an underlying ischemic heart disease. Because ischemic heart disease is not prevalent in the young, in these cases, SCD may often be related to a familial, inherited, or genetic disease.2,3
A careful cardiac investigation during the autopsy can provide a definitive diagnosis in structural heart diseases, be it by the identification of a culprit coronary lesion or heart muscle disease, which explain the episode. However, a significant number of SCD cases remain unexplained after a comprehensive medicolegal investigation. These autopsies are usually labeled as negative, natural, or arrhythmogenic. In these negative autopsies, especially in the young, a genetic disease causing an electrical disturbance (ion channelopathy) should be immediately suspected. Because of the familial nature of the disease, the identification of these genetic defects carry important implications for relatives who are at potential risk of also having a fatal cardiac condition.4,5
Several mutations in genes that encode cardiac ion channels and structural proteins have already been associated with fatal and nonfatal arrhythmias, although the basic mechanisms have yet to be fully elucidated.6 Genetic testing has therefore become a new tool in clinical cardiology. As such, when the clinical diagnosis is clear, genetic testing may help confirm the disease and identify genetic carriers in the family. A genetic study may even be of help in borderline phenotypes, because sometimes it becomes the only means of diagnosing a potentially fatal disease. However, when the clinical phenotype is not clear (i.e., sudden unexplained death), genetic testing continues to be a fishing expedition, which can only be performed for research but not for clinical purposes, because it is not cost effective.
Despite some significant overlaps, inherited cardiac diseases associated with SCD can be classified into two broad groups (Fig. 1)7:

Arrhythmogenic factors. Arrhythmogenesis depends on the critical interaction between environmental and genetic factors. The equilibrium prevents the arrhythmias. Alteration of only one may already induce arrhythmogenesis.
-
a
Primary electrical diseases, or channelopathies, in which the arrhythmogenic substrate is found in the electrical properties of the heart, and
-
b
Cardiomyopathies, in which structural abnormalities such as hypertrophy, dilatation, or fatty and/or fibrotic infiltration, are responsible for inducing the arrhythmia.
In this review, we focus on the genetic basis of the primary electrical diseases or ion channelopathies.
ION CHANNEL DISEASES
Ion channels are integral membrane proteins that are responsible for ion conduction across the cell membrane. Ion channel subunits are multimeric proteins formed by different subunits, which are usually encoded by separate genes. Ion channel subunits are formed by pore-forming α-subunits, which mediate ion currents, and regulatory β-subunits. In the heart, as in other excitable tissues, the generation of the action potential results from the complex balance of several ion currents. When this balance is disrupted by the altered current generated by a faulty ion channel (due to a genetic mutation), there may be a risk for electrical instability (arrhythmias in the heart or epilepsy in the brain). Channelopathy is thus defined as an inherited syndrome caused by mutations in genes encoding for ion channels, their subunits, or associated proteins.8–13
Several mutation-associated cardiac diseases, affecting sodium (Na+), potassium (K+), or calcium (Ca2+) ion currents, have been described affecting either the generation of the action potential or calcium homeostasis. Cardiac channelopathies may be clinically identified only by the presence of some characteristic electrocardiographic (ECG) abnormalities.14–16 These are not always present in genetic carriers, because the diseases often have a low penetrance.17–20 In these situations, genetic studies may be key to providing a lead toward an etiology for the unexplained symptoms.21
Diseases associated with sodium channel dysfunction
The α-subunit of the cardiac isoform of the sodium channel is known as Nav1.5 and is encoded by the SCN5A gene. Nav1.5 is a membrane protein that contains 2015 or 2016 residues (depending on the splice variant) for an approximate molecular weight of 227 kDa. It consists of four homologous domains, known as DI to DIV, joined by so-called linkers. These three linkers in addition to the N-terminus and the C-terminus of the protein are cytoplasmic. Each domain, DI to DIV, contains six transmembrane helices (namely S1–S6) linked by intracellular or extracellular loops. S5 and S6 in each of the domains form the pore-lining helices, and the extracellular loop between them is the longest among extracellular segments of Nav1.5.
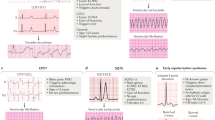
Malfunctioning of cardiac Na+ channels is the cause of some primary arrhythmia syndromes. Among others, four diseases have been associated to genetic mutations that affect Na+ channel function: Brugada syndrome (BrS), Lev-Lenègre syndrome (familial progressive conduction disease), long QT syndrome (LQTS), and familial atrial fibrillation (AF). Certain mutations can give rise to different phenotypes with enhanced or reduced channel function and even combinations of phenotypes8 (Table 1).
Brugada syndrome
BrS is a hereditary disease responsible for ventricular fibrillation and SCD in the young. The disease is characterized by the presence of right ventricular conduction abnormalities and coved-type ST-segment elevation in the anterior precordial leads (V1–V3). Patients with BrS have a structurally normal heart, although minor structural alterations have been described in some cases.17–20 The prevalence of BrS is estimated to be ∼35/100,000 person/year. Although the mean age of onset of events is ∼40 years, SCD can affect individuals of any age, particularly men (75%). Of those patients affected, 20–50% has a family history of SCD.
The disease exhibits an autosomal dominant pattern of transmission and variable penetrance.13,22 Most mutations occur in genes related to Na+ channels, although other channels may also be implicated in BrS.23 Between 20 and 25% of patients affected by BrS have mutations in the SCN5A gene,24 which encodes the α-subunit of the cardiac Na+ channel. To date, there are >200 SCN5A BrS-related mutations described. These mutations lead a loss of Na+ channel function through several mechanisms.8,12,13,24–28 Modulation of the phenotype may be related to the presence of environmental factors (i.e., inducers like fever, or medications) or additional genetic factors. A combination of two BrS mutations, each of which can produce functional but biophysically defective sodium channels, is associated with a more severe phenotype.29 Similarly, several promoter polymorphisms have been described in SCN5A, which may also affect Na+ channel function. A haplotype of six polymorphisms in the SCN5A promoter has been functionally linked to a reduced expression of the sodium current. This variant was found among patients of Asian origin and could explain the different clinical phenotypes and increased incidence of BrS in certain geographic regions in the Southeast Asia.30–33
The second gene linked to BrS is GPD1-L, a Na+ channel-associated mutation also responsible of sudden death (SD) in neonates.34 GPD1-L encodes for a glycerol-3-phosphate dehydrogenase-like protein of 351 amino acids. It has been shown that mutations in GPD1-L impair the surface membrane expression of the ion channel and reduce inward Na+ current.35 Other recently described minor contributors to BrS are mutations in the genes SCN1B (sodium channel beta-1 subunit) and SCN3B (beta-3 subunit of the cardiac sodium channel).36,37
Lev-Lenègre syndrome
Lev-Lenègre syndrome is a progressive cardiac conduction disease characterized by gradually developed ventricular fibrosis of the conduction system, which may lead to arrhythmias or asystolia.38 The first locus for the disease was reported in 1995, on chromosome 19q13.2–13.3.39 In 1999, the first mutations causing the disease were associated with SCN5A.24 Mutations in SCN5A lead to a reduction in Na+ current, reducing the velocity of the impulse conduction.11
Long QT syndrome
LQTS is a cardiac channelopathy characterized by a QT segment prolongation, responsible for ventricular tachyarrhythmias with associated episodes of syncope and SD.40 LQTS is one of the leading causes of SD among young people. It can be congenital or acquired, generally in association with drugs and electrolyte imbalance (hypokalemia, hypocalcemia, and hypomagnesemia).41 The clinical presentation can be variable, ranging from asymptomatic patients to episodes of syncope and SD caused by ventricular tachyarrhythmias (torsade de pointes) in a structurally normal heart.40,42 Prolongation of the QT interval may arise because of a decrease in the K+ repolarization currents or to an inappropriate delay in the entry of Na+ into the myocyte.
Until now, 12 different types of LQTS have been described, most of them associated to K+ channel disorders (potassium channel encoding genes). However, a 10–15% of the LQTS cases are related to mutations in SCN5A (Type 3 LQTS).38 Patients with Type 3 LQTS present arrhythmias associated with bradycardia and symptoms at rest, especially during sleep. Type 3 LQT patients do not respond as well to β-blockers, and the presence of a long QT3 mutation is a sign of high risk for events. LQTS Type 10 is caused by a mutation in the SCN4B gene, which encodes the β4-subunit of the cardiac Na+ channel (Nav4b).43 This mutation induces a positive change in the inactivation of Na+ channels, which increases Na+ current and delays the repolarization in a similar fashion to Type 3 LQTS.
Atrial fibrillation
AF is defined as an unpredictable activation of the atria, characterized by irregular fibrillatory ECG waves causing an irregular ventricular response, which manifests clinically as an irregular pulse. AF is one of the most common and yet challenging arrhythmias encountered in clinical practice. The prevalence of 1% among the general population increases to 10% among individuals older than 80 years and it is responsible for over one third of all cardioembolic episodes.44 Similarly, environmental factors have been reported to be particularly important in the onset and course of the condition.45,46
Although familial forms had remained mostly unknown, the identification, in 1997, of a genetic locus causing familial AF47 defined AF as a genetic disease with an autosomal dominant pattern of inheritance and initiated a deeper research into the understanding of the pathophysiology of this inherited form of the arrhythmia.
To date, most of the genes associated to AF encode K+ channel proteins (KCNQ1, KCNE2, KCNE3, KCNA5, KCNJ2, and KCNH2).48 However, AF has also been associated with mutations in SCN5A6,49 and recently to sodium channel beta-subunits SCN1B and SCN2B,50 in patients with AF. These findings further support the hypothesis that decreased sodium current enhances AF susceptibility. A combined phenotype of long QT3 and AF has also been described, obviously caused by a gain of function mutation.51
Diseases associated with potassium channel dysfunction
The potassium channels most relevant to heart diseases are the slowly activating delayed rectifier cardiac K+ channel and the voltage-gated inwardly rectifying K+ channel. The α-subunits of the cardiac isoforms of these two potassium channels are known as KvLQT1 and KCNH2, respectively. Potassium channels allow repolarization currents to counteract the preceding depolarization process.27,52 Mutations in the genes that encode the K+ channels may give rise to three types of disease: LQTS, short QT syndrome (SQTS), and AF (Table 1).
Long QT syndrome
LQTS is a cardiac channelopathy mainly caused by repolarization abnormalities related to K+ channels. Several mutations have been reported in K+ channels, all of which are related to a loss of function.42 Fifty to sixty percent of the clinical cases of LQTS are associated to mutations located on six different K+ channel genes.53,54 Inheritance can follow an autosomal dominant (Romano-Ward syndrome) or recessive (Jervell and Lange-Nielsen syndrome) transmission pattern.55
The most common form of the LQTS, known as Type 1 LQTS,56,57 is caused by mutations in KCNQ1. The gene product of KCNQ1, namely KvLQT1, encodes for the α-subunit of the slowly activating delayed rectifier cardiac K+ channel, IKs. To date, 40–50% of the cases of prolonged QT interval are associated to mutations in KCNQ1.58,59 KvLQT1 forms a complex with the protein encoded by the KCNE1 gene (MinK, β-subunit of the K+ channel), which regulates IKr and IKs activity.60,61 Five mutations have been reported in the KCNE1 gene,62 which induce 2–5% of the cases (i.e., LQTS Type 5).
KCNH2 (human-ether-a-go-go-related gene) encodes for the α-subunit of a voltage-gated inwardly rectifying K+ channel that mediates the rapidly activating component of IKr in the heart. Mutations in KCNH2 lead to loss of function of the K+ channel and account for 35–45% of cases of the so-called Type 2 LQTS.63–65 KCNH2 associates with KCNE2, a gene which encodes MiRP1, β-subunit of the rapidly activating component of IKr. KCNE2 causes Type 6 LQTS, a very rare syndrome (<1%) which leads to loss of channel function.66
LQTS Type 7, or Tawil-Anderson syndrome, is caused by loss of function mutations in KCNJ2. This gene encodes IK1 (Kir2.1), an inward rectifying K+ channel.67 The incidence is very low (only eight natural variants have been described to date) and rarely associated with syncopes or SD68; however, episodes of polymorphic or bidirectional tachycardia may occur.69
Short QT syndrome
Short QT syndrome is a clinically malignant entity2 characterized by a short QT interval (<330 milliseconds), with tall, symmetric and peaked T waves leading to ventricular arrhythmias and SD.70 It may cause SD in neonates, because clinical manifestations may appear early in life. Most patients have a family history of SD with an autosomal dominant pattern of transmission and a high penetrance.71
Mutations of three K+ channels have been related to SQTS due to enhanced function and, therefore, shortened repolarization. Type 1 SQTS has been associated with the KCNH2 gene (human-ether-a-go-go-related gene), which induces a fast activation of K+ currents, with enhanced IKr function and shortened ventricular action potentials.15 The mutation described may present with AF in some families.72
The SQTS Type 2 has been linked to mutations in KCNQ1.71–73 The mutation enhances the function of the K+ channel, leading to a shortening of the action potential with AF.72 Curiously enough, a rare entity caused by a KCNQ1 mutation has been described, which is manifested as bradycardia in utero, and short QT and AF in the neonatal period.73
The SQTS Type 3 is caused by a mutation in KCNJ2 gene, which accelerates the Phase 3 of the cardiac action potential.74 This form of short QT has a characteristic ECG pattern, described as an abrupt and sharp termination of the T wave.
Atrial fibrillation
AF is the most common arrhythmia observed in clinical practice. Several loci have been mapped and most of them encode for subunits of potassium channels.48 In 2003, the identification of a locus on chromosome 11 in a four-generation family and the subsequent identification of KCNQ1 as responsible for the disease provided the first link of the genetic form of the arrhythmia with an ion channelopathy.45 The analysis of KvLQT1 (KCNQ1) identified a missense mutation resulting in the amino acid change S140G. Electrophysiological studies revealed a gain of function in Iks current when the mutated channel was expressed with the β-subunits MinK and MirP1. This gain of function explained well the shortening of the action potential duration and effective refractory period, which are thought to be the culprits of the disease.45 The role of KCNQ1 in AF has recently advanced,75 with identification of a mutation in KCNQ1, (R14C) in an interesting family with AF. The mutant protein only showed increase IKs upon cell stretch with a hypotonic solution opening a new and stimulating debate on the role of genetic-environmental interaction in the development of the disease. In 2005, Hong et al.,73 described a gain of function mutation in codon 141, responsible for a severe form of AF, in utero and SQTS.
Defects in genes encoding for potassium currents were further confirmed responsible for AF with the identification of gain of function mutations in KCNE2,76 in KCNH2 (causing AF and short-QT-syndrome)72 and in Kir2.1.77 A genetic defect has been described in KCNE378; however, the functional analysis did not demonstrate a different biophysical effect caused by the mutant genetic defect, indicating that it could be a rare polymorphism. In summary, the biophysical findings therefore indicated a role of gain-of-function mutations in potassium channels in AF highlighting the pathophysiological role of shortened atrial action potentials. When Olson et al.79 described a loss-of-function mutation in KCNA5, the gene that encodes Kv1.5, the debate became more stimulating because it initiated the hypothesis that a prolongation of the action potential can also be a basic mechanism for development of AF.
Diseases associated with calcium channel dysfunction
The voltage-gated calcium channel subunit alpha, Cav1.2, encoded by CACNA1C, is a 2221 amino acid protein with a molecular weight of 250 kDa. It is a membrane protein with a similar topology to the α-subunit of the sodium channel: it consists of four homologous domains (DI to DIV), joined by cytosolic linkers. Each domain also contains six transmembrane helices. On the other hand, the Ryanodine receptor 2, encoded by RyR2, and the calsequestrin 2, encoded by the RyR2 and CASQ2, respectively, locate at the sarcoplasmic reticulum. The Ryanodine receptor 2 is a huge protein of 4965 residues that has 12 transmembrane helices and a variety of domains, whereas CASQ2 is a small protein of 399 residues (46 kDa).
Calcium ions are involved in phase 2 of cardiac action potential and also increase the output of calcium from the sarcoplasmic reticulum, which functions primarily as a storehouse of calcium in the skeletal muscle, to trigger cardiac contraction. The role of calcium channels in inherited arrhythmia syndromes has only recently been demonstrated.80 To date, a Ca2+ channel dysfunction can produce three different diseases—a combination of BrS with shorter than normal QT interval, Timothy syndrome (TS), and Polymorphic Ventricular Tachycardia (Table 1).
Combination of BrS and shorter than normal QT intervals
Mutations in L-type Ca2+ channel (CACNA1C) or its β2b-subunit (CACNB2b) have been reported in BrS patients associated with a shorter than normal QT-interval (>330 milliseconds but <360 milliseconds in male probands), which is to be expected in the presence of decreased calcium inward current. The diseases associated with mutations in CACNA1C and CACNB2b follow an autosomal dominant pattern of transmission. These mutations (A39V and G490R in CACNA1C and S481L in CACNB2b) induce a loss of calcium channel function, which in the case of the A39V mutant is most likely caused by defective trafficking to the cell membrane.81,82
Timothy syndrome
TS is an uncommon disease characterized by the presence of a multiorgan dysfunction, which includes severe arrhythmias with the highest associated mortality.83 TS is also named LQT Type 8,84 caused by mutations in CACNA1C.85 Two mutations have been reported in CACNA1C gene, which induce a gain of function with an altered ICa with a loss of voltage-dependent channel leading to a prolongation of action potential and QT interval.85 The effect of the TS mutation on other L-channel gating mechanisms has yet to be investigated.83,86–88 Some groups explore mechanisms by which beta-adrenergic stimulation modulates arrhythmogenesis in order to identify potential targets for antiarrhythmic therapy.89
Polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a familial arrhythmogenic disorder characterized by the presence of SCD sometimes associated with bidirectional polymorphic ventricular tachycardia. Patients show a normal resting ECG although adrenergic stimulation (vigorous exercise, fear) may induce syncope and/or SCD (30–50% by the age of 20–30 years) in a structurally normal heart.90–92
Two genetic forms have been identified responsible for CPVT. First, an autosomal dominant form caused by mutations (>120 have been identified to date) in the RyR2 gene (1q42–q43), encoding the ryanodine receptor (CPVT Type 1).93 Second, a recessive form, caused by mutations in calsequestrin (CASQ2) (CPVT Type 2), located on 1p13–p21.94–96 These genes are key players in the storage and calcium release from the sarcoplasmatic reticulum to the cytosol. Both genes are therefore implicated in the mediation of excitation-contraction coupling. In general, mutations lead to increased intracellular Ca2+, leading to late depolarizations which predispose to arrhythmias.95
Diseases associated with dysfunction of ion channel-associated proteins
Mutations in proteins involved in the trafficking of ion channels into the cell membrane have also been identified as key factors leading to cardiac arrhythmias. For instance, the ANK2 gene encodes the ankyrin-B protein, which attaches membrane proteins such as the Na+/K+ ATPase pump, the Na+/Ca2+ exchanger, and the inositol triphosphate receptor, to cytoskeletal structures.97 Five mutations in ANK2 have been described as responsible for LQT Type 4, characterized by fatal ventricular arrhythmias.
Caveoline-3 (Cav3) is a scaffolding protein implicated in membrane trafficking and positioning of the ion channels in the sarcoplasmatic membrane. Five reported mutations in Cav3 are responsible for an enhancement of Na+ channel function, leading to LQT Type 9.98
Syntrophins are cytoplasmic submembranous proteins that are components of the dystrophin associated protein complex.99 The cytoskeletal protein syntrophin-α1 (SNTA1) is known to interact with the cardiac sodium channel (Nav1.5). The reported mutations in STNA1 cause gain-of function of Nav1.5 with increased late INa, similar to the LQT3.100,101
Atrial natriuretic peptide precursor encodes atrial natriuretic peptide. Atrial natriuretic peptide modulates ionic currents in cardiac myocytes and can play a role in shortening of the atrial conduction time, which could be a potential substrate for atrial reentrant arrhythmias. In 2008, Hodgson-Zingman et al.102 identified a frame shift mutation in atrial natriuretic peptide precursor in a large family with AF.
The latest gene to be linked to AF is the one identified by Zhang et al.103 The clinical phenotype was characterized by a neonatal onset, with autosomal recessive inheritance. They identified a mutation in NUP155, which encode a member of the nucleoporins. Although still unknown, the mechanism by which NUP155 may be associated with AF could be related to modulation of calcium handling proteins and ion channel and expression of its possible target genes, like HSP70. This gene is located in 5p13 and had been also associated with SD in the family.
GENETIC TESTING AND PERSONALIZED MEDICINE
The relevance of the sequencing of the human genome in the understanding of a wide range of diseases cannot be understated. In particular, genetics and genetic-molecular technology have had a great impact in research of cardiovascular diseases. In these past years, thanks to research in genetics, we have identified several disease-associated genes. Obviously, the research findings in the probands have clinical implications not only for them but also for their family members. The classic paradigm of dealing with a patient with signs and symptoms for a disease has been changed with the advent of genetics, as now we are able to identify individuals at risk (genetic carriers), usually family members, who have just a genetic variation. As a result, a new area has emerged in clinical cardiology, cardiovascular genetics, which has to deal with a new level of complexity, not with patients but with families.
Because of the inherited scenario, it is imperative that the family is approached and studied after the death of the loved one from a suspected genetic disease. The previous knowledge of an electrical disease in the proband or the family member may help find an etiology for the sudden death, may provide closure to the family, and may enable the adoption of protective measures to prevent a second SCD. Therefore, clinical investigation is important in this situation. When the clinical tools have not been sufficient, genetic investigation and the identification of a potential disease-related mutation may be extremely important; the identification of the mutation in a family member will confirm that he or she is also at risk for the genetic disease and may receive preventive therapies. Those without the familial mutation are spared from that familial disease. This is critical and useful information.
It is also obvious that the suspicion of a familiar disease will bring anxiety and concern to the families who are learning that they may be also at risk while still mourning. Therapeutic decisions in asymptomatic genetic carriers are difficult, and physicians, patients, and family members will have to cope with complex new information. Multidisciplinary teams that incorporate cardiologists, geneticists, and genetic counselors are crucial to answer potential concerns, to be expeditious in the clinical and genetic investigations, and to approach the several unknowns that still remain. These teams have been recently organized through clinical cardiovascular genetics centers and are available in any major university center.
References
Priori SG, Aliot E, Blomstrom-Lundqvist C, et al. Task force on sudden cardiac death, European Society of Cardiology. Europace 2002; 4: 3–18.
Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome?. Cardiology 2000; 94: 99–102.
Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006; 114: e385–e484.
Chugh SS, Kelly KL, Titus JL . Sudden cardiac death with apparently normal heart. Circulation 2000; 102: 649–654.
Chugh SS, Senashova O, Watts A, et al. Postmortem molecular screening in unexplained sudden death. J Am Coll Cardiol 2004; 43: 1625–1629.
Ellinor PT, Nam EG, Shea MA, Milan DJ, Ruskin JN, MacRae CA . Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 2008; 5: 99–105.
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816.
Grant AO, Carboni MP, Neplioueva V, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 2002; 110: 1201–1209.
Benson DW, Wang DW, Dyment M, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 2003; 112: 1019–1028.
Bennett PB, Yazawa K, Makita N, George AL Jr, Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995; 376: 683–685.
Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001; 409: 1043–1047.
Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA, Balser JR . Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res 2000; 86: E91–E97.
Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293–296.
Roden DM, Lazzara R, Rosen M, Schwartz PJ, Towbin J, Vincent GM . Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation 1996; 94: 1996–2012.
Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109: 30–35.
Hong K, Brugada J, Oliva A, et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation 2004; 110: 3023–3027.
Papavassiliu T, Wolpert C, Fluchter S, et al. Magnetic resonance imaging findings in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2004; 15: 1133–1138.
Coronel R, Casini S, Koopmann TT, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005; 112: 2769–2777.
Frustaci A, Priori SG, Pieroni M, et al. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 2005; 112: 3680–3687.
Frustaci A, Russo MA, Chimenti C . Structural myocardial abnormalities in asymptomatic family members with Brugada syndrome and SCN5A gene mutation. Eur Heart J 2009; 30: 1763.
Alders M, Koopmann TT, Christiaans I, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet 2009; 84: 468–476.
Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet 2002; 11: 337–345.
Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation 2002; 105: 707–713.
Schott JJ, Alshinawi C, Kyndt F, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 1999; 23: 20–21.
Wang DW, Makita N, Kitabatake A, Balser JR, George AL Jr, Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ Res 2000; 87: E37–E43.
Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85: 803–809.
Rivolta I, Abriel H, Tateyama M, et al. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem 2001; 276: 30623–30630.
Clancy CE, Rudy Y . Na(+) channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation 2002; 105: 1208–1213.
Cordeiro JM, Barajas-Martinez H, Hong K, et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation 2006; 114: 2026–2033.
Poelzing S, Forleo C, Samodell M, et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 2006; 114: 368–376.
Baron RC, Thacker SB, Gorelkin L, Vernon AA, Taylor WR, Choi K . Sudden death among Southeast Asian refugees. An unexplained nocturnal phenomenon. JAMA 1983; 250: 2947–2951.
Bezzina CR, Shimizu W, Yang P, et al. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation 2006; 113: 338–344.
Ito H, Yano K, Chen R, He Q, Curb JD . The prevalence and prognosis of a Brugada-type electrocardiogram in a population of middle-aged Japanese-American men with follow-up of three decades. Am J Med Sci 2006; 331: 25–29.
Van Norstrand DW, Valdivia CR, Tester DJ, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 2007; 116: 2253–2259.
London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007; 116: 2260–2268.
Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 2008; 118: 2260–2268.
Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the b3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2009; 2: 270–278.
Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995; 80: 805–811.
Brink PA, Ferreira A, Moolman JC, Weymar HW, van der Merwe PL, Corfield VA . Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation 1995; 91: 1633–1640.
Moss AJ, Robinson JL . Clinical aspects of the idiopathic long QT syndrome. Ann N Y Acad Sci 1992; 644: 103–111.
Zipes DP . The long QT interval syndrome. A Rosetta stone for sympathetic related ventricular tachyarrhythmias. Circulation 1991; 84: 1414–1419.
Roden DM . Cellular basis of drug-induced torsades de pointes. Br J Pharmacol 2008; 154: 1502–1507.
Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 2007; 116: 134–142.
Anderson ME . When will we know enough to treat atrial fibrillation?. Heart Rhythm 2007; 4: 750–751.
Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003; 299: 251–254.
Chen S, Zhang L, Bryant RM, et al. KCNQ1 mutations in patients with a family history of lethal cardiac arrhythmias and sudden death. Clin Genet 2003; 63: 273–282.
Brugada R, Tapscott T, Czernuszewicz GZ, et al. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med 1997; 336: 905–911.
Tsai CT, Lai LP, Hwang JJ, et al. Molecular genetics of atrial fibrillation. J Am Coll Cardiol 2008; 52: 241–250.
Darbar D, Kannankeril PJ, Donahue BS, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008; 117: 1927–1935.
Watanabe H, Darbar D, Kaiser DW, et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol 2009; 2: 268–275.
Benito B, Brugada R, Perich RM, et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 2008; 5: 1434–1440.
Ravens U, Cerbai E . Role of potassium currents in cardiac arrhythmias. Europace 2008; 10: 1133–1137.
Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 2004; 292: 1341–1344.
Westenskow P, Splawski I, Timothy KW, et al. Compound mutations: a common cause of severe long-QT syndrome. Circulation 2004; 109: 1834–1841.
Jervell A, Lange-Nielsen F . Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957; 54: 59–68.
Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G . K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996; 384: 78–80.
Olszak-Waskiewicz M, Dziuk M, Kubik L, Kaczanowski R, Kucharczyk K . Novel KCNQ1 mutations in patients after myocardial infarction. Cardiol J 2008; 15: 252–260.
Aizawa Y, Ueda K, Scornik F, et al. A novel mutation in KCNQ1 associated with a potent dominant negative effect as the basis for the LQT1 form of the long QT syndrome. J Cardiovasc Electrophysiol 2007; 18: 972–977.
Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007; 115: 2481–2489.
Bianchi L, Shen Z, Dennis AT, et al. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum Mol Genet 1999; 8: 1499–1507.
McDonald TV, Yu Z, Ming Z, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 1997; 388: 289–292.
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT . A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995; 80: 795–803.
Sanguinetti MC, Jiang C, Curran ME, Keating MT . A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995; 81: 299–307.
Sanguinetti MC, Curran ME, Spector PS, Keating MT . Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci USA 1996; 93: 2208–2212.
Shimizu W, Moss AJ, Wilde AA, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol 2009; 54: 2052–2062.
Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999; 97: 175–187.
Tsuboi M, Antzelevitch C . Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm 2006; 3: 328–335.
Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001; 105: 511–519.
Tawil R, Ptacek LJ, Pavlakis SG, et al. Andersen's syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol 1994; 35: 326–330.
Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43: 1494–1499.
Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109: 2394–2397.
Hong K, Bjerregaard P, Gussak I, Brugada R . Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol 2005; 16: 394–396.
Hong K, Piper DR, Diaz-Valdecantos A, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res 2005; 68: 433–440.
Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96: 800–807.
Otway R, Vandenberg JI, Fatkin D . Atrial fibrillation—a new cardiac channelopathy. Heart Lung Circ 2007; 16: 356–360.
Yang Y, Xia M, Jin Q, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet 2004; 75: 899–905.
Xia M, Jin Q, Bendahhou S, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 2005; 332: 1012–1019.
Zhang DF, Liang B, Lin J, Liu B, Zhou QS, Yang YQ . [KCNE3 R53H substitution in familial atrial fibrillation.]. Chin Med J (Engl) 2005; 118: 1735–1738.
Olson TM, Alekseev AE, Liu XK, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet 2006; 15: 2185–2191.
Wilde AA . Channelopathies in children and adults. Pacing Clin Electrophysiol 2008; 31( suppl 1): S41–S45.
Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115: 442–449.
Antzelevitch C . Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4: 964–972.
Yarotskyy V, Gao G, Peterson BZ, Elmslie KS . The Timothy syndrome mutation of cardiac CaV1.2 (L-type) channels: multiple altered gating mechanisms and pharmacological restoration of inactivation. J Physiol 2009; 587( Pt 3): 551–565.
Lehnart SE, Ackerman MJ, Benson DW Jr, et al. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation 2007; 116: 2325–2345.
Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19–31.
Zhu ZI, Clancy CE . L-type Ca2+ channel mutations and T-wave alternans: a model study. Am J Physiol Heart Circ Physiol 2007; 293: H3480–H3489.
Sicouri S, Glass A, Ferreiro M, Antzelevitch C Transseptal dispersion of repolarization and its role in the development of torsade de pointes arrhythmias [published online ahead of print November 10, 2010]. J Cardiovasc Electrophysiol Doi: 10.1111/j.1540-8167.2009.01641.x.
Sicouri S, Timothy KW, Zygmunt AC, et al. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: effects of ranolazine. Heart Rhythm 2007; 4: 638–647.
Sung RJ, Wu YH, Lai NH, et al. Beta-adrenergic modulation of arrhythmogenesis and identification of targeted sites of antiarrhythmic therapy in Timothy (LQT8) syndrome: a theoretical study. Am J Physiol Heart Circ Physiol 2010; 298: H33–H44.
Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003; 113: 829–840.
Coumel P . Catecholaminergic polymorphic ventricular tachyarrhythmias in children. Card Electrophysiol Rev 2002; 6: 93–95.
Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P . Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995; 91: 1512–1519.
Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol 2009; 54: 2065–2074.
Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001; 103: 485–490.
Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103: 196–200.
Lahat H, Eldar M, Levy-Nissenbaum E, et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation 2001; 103: 2822–2827.
Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003; 421: 634–639.
Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 2006; 114: 2104–2112.
Guglieri M, Magri F, Comi GP . Molecular etiopathogenesis of limb girdle muscular and congenital muscular dystrophies: boundaries and contiguities. Clin Chim Acta 2005; 361: 54–79.
Wu G, Ai T, Kim JJ, et al. Alpha-1-syntrophin mutation and the long-QT syndrome: a disease of sodium channel disruption. Circ Arrhythm Electrophysiol 2008; 1: 193–201.
Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci USA 2008; 105: 9355–9360.
Hodgson-Zingman DM, Karst ML, Zingman LV, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med 2008; 359: 158–165.
Zhang X, Chen S, Yoo S, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 2008; 135: 1017–1027.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Campuzano, O., Beltrán-Álvarez, P., Iglesias, A. et al. Genetics and cardiac channelopathies. Genet Med 12, 260–267 (2010). https://doi.org/10.1097/GIM.0b013e3181d81636
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181d81636
Keywords
This article is cited by
-
Unexplained cardiac arrest: a tale of conflicting interpretations of KCNQ1 genetic test results
Clinical Research in Cardiology (2018)
-
Precision medicine approach to genetic cardiomyopathy
Herz (2017)
-
Genetic Predispositions to Heart Failure
Current Cardiovascular Risk Reports (2016)
-
Determining the Pathogenicity of Genetic Variants Associated with Cardiac Channelopathies
Scientific Reports (2015)
-
Negative autopsy and sudden cardiac death
International Journal of Legal Medicine (2014)
