
Abstract
Purpose
To investigate phenotypic variability in terms of best-corrected visual acuity (BCVA) in patients with Stargardt disease (STGD) and confirmed ABCA4 mutations.
Methods
Entire coding region analysis of the ABCA4 gene by direct sequencing of seven patients with clinical findings of STGD seen in the Retina Clinics of Southampton Eye Unit between 2002 and 2011.
Phenotypic variables recorded were BCVA, fluorescein angiographic appearance, electrophysiology, and visual fields.
Results
All patients had heterozygous amino acid-changing variants (missense mutations) in the ABCA4 gene.
A splice sequence change was found in a 30-year-old patient with severly affected vision. Two novel sequence changes were identified: a missense mutation in a mildly affected 44-year-old patient and a frameshift mutation in a severly affected 34-year-old patient.
Conclusion
The identified ABCA4 mutations were compatible with the resulting phenotypes in terms of BCVA.
Higher BCVAs were recorded in patients with missense mutations. Sequence changes, predicted to have more deleterious effect on protein function, resulted in a more severe phenotype.
This case series of STGD patients demonstrates novel genotype/phenotype correlations, which may be useful to counselling of patients. This information may prove useful in selection of candidates for clinical trials in ABCA4 disease.
Similar content being viewed by others

Introduction
The ABCA4 gene produces an ATP-binding cassette superfamily transmembrane protein expressed in retinal photoreceptors and involved in the transport of substrates across membranes. It is a large gene consisting of 50 exons. Missense mutations in this gene were first identified as causing Stargardt disease (STGD) or fundus flavimaculatus (FFM).1, 2
Subsequently, it was appreciated that mutations that resulted in truncation or severe misfolding of the ABCA4 protein led to more severe retinal phenotypes of retinitis pigmentosa and cone-rod dystrophy. Intrafamilial variability of the phenotype in STGD has also been reported. This suggests that other genes or the environment affect the phenotypic expression of a given ABCA4 genotype.1
Case reports
Analysis was performed for the entire coding region of the ABCA4 gene by direct sequencing of seven patients with clinical findings of STGD, seen in the Retina Clinics of Southampton Eye Unit between 2002 and 2011. The whole coding sequence of the ABCA4 gene was sequenced at the Netherlands Institute for Neuroscience in Amsterdam.
Phenotypic variables recorded were best-corrected visual acuities, fluorescein angiographic appearance, electrophysiology, and visual fields. We focused on best-corrected visual acuity (BCVA), which is an easy to access and useful marker of disease progression and prognosis. We estimated disease duration based on the date of first-time diagnosis of the disease by a retina specialist.
This was a retrospective study and we did not ask for institutional approval as the investigations were part of patients’ routine clinical care without any additional interventions occurring.
Case 1
A 44-year-old man presented with a gradual, relatively recent decreased visual acuity in the right eye (logMar BCVA OD: 032, OS: 0.02), no previous ocular or medical history, and no known family history of an ocular disease. On fundus examination, there was some pigment mottling in both maculae and ‘fish-tail’-like retinal flecks with a central distribution mainly in the right eye (Figure 1). Visual fields revealed a central relative scotoma in the right eye but the test was normal for the left eye. The fluorescein angiogram displayed a characteristic ‘dark choroid’ in the right eye and a patchy pattern of choroidal hyperfluorescence in the left eye. Full-field electroretinogram (ERG) was normal in both eyes, whereas the pattern ERG (PERG) was reduced in both eyes, with a lower PERG amplitude in the right eye. Analysis of the entire coding region of the ABCA4 gene revealed a nucleotide change (missense) mutation, 191C>T, which has not been reported in the literature before and confirmed the diagnosis of STGD.

Coloured fundus photographs and fluorescein angiography images of both eyes of case 1 where a novel missense mutation, 191C>T, was identified.
Case 2
A 29-year-old woman presented with a vague description of visual problems, mainly complaining of a decreased ability to focus while reading and a recently increasing difficulty to adapt in the dark but normal visual acuity in both eyes (logMar BCVA OD/OS: 0.02), and no other ocular or medical history or a family history of an ocular disease. Fundus examination was unremarkable as well as visual fields and fluorescein angiogram were rejected by the patient. However, electrophysiology testing enhanced the suspicion of a possible Stargardt’s diagnosis as PERG responses were diminished in both eyes. Analysis of the ABCA4 gene revealed two, known to cause STGD, heterozygous amino acid-changing variants, 1805G>A and 6079C>T.
Case 3
A 34–year-old woman presented with a bilateral markedly decreased vision (logMar BCVA OD:0.93, OS:0.95), which according to the patient started deteriorating a year before presentation and had recently become even worse. There was no previous ocular history or a remarkable medical history and no family history of an ocular disease either. Fundus examination showed bilateral atrophic lesions of the macula accompanied by yellow-white stellate flecks at the level of the retinal pigment epithelium. Atrophic lesions and flecks were also extending to the mid-periphery of both retinae. Visual fields revealed a bilateral absolute central scotoma and relative paracentral scotomas as well in both eyes. PERG was significantly reduced, while scotopic and photopic amplitudes were also lower than normal. Analysis of the ABCA4 gene revealed a missense mutation, 5882G>A, recorded in the literature before as well as a frameshift mutation, 6709insA, that in the best of our knowledge has not been previously reported.
Cases 4–7
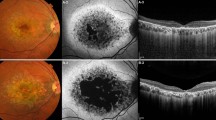
Four of the patients included in this case series were diagnosed with STGD years ago based on clinical findings and ancillary tests; however, the diagnosis was never genetically confirmed (Table 1). The patients’ age ranged from 34 to 55 years. Three of them were men and one was woman. Disease duration varied from 8 to 22 years. BCVA was significantly reduced in both their eyes and ranged between 0.22 and 1.34 logMar. Visual fields, FFA, and electrophysiological tests were not all consistently performed in all patients but we did analyse the coding region of the ABCA4 gene, and all of them were found to have in heterozygous form amino acid-changing variants. A splice sequence change was additionally found in a 30-year-old patient with severly affected vision (Figure 2).

Coloured fundus photographs of both eyes of case 6 where a splice sequence change, 5018+2T>C, previously reported, was identified. The progress of clinical findings between 2007 (images above) and 2012 (images below) is clearly displayed.
Discussion
There are 700 disease-associated variants for the ABCA4 gene and more than half of them have been detected only once.9 As ABCA4-associated diseases are recessive, it is common for a patient to be the only affected member in a family. It is therefore difficult to determine the pathogenicity of rare variants. Hence, it is useful to report additional patients to help confirm the pathogenicity of rare mutations. Correct molecular diagnosis and genotype/phenotype correlations remain a crucial step towards understanding the broad spectrum of ABCA4 retinopathies. In addition, recruitment of patients in clinical trials that aim to investigate effectiveness and safety of treatment options require molecular confirmation of the disease. Correlation of specific genotypes with phenotypic progress will also aid the design and assessment of patients in clinical trials.
Genotype/phenotype correlation in our small case series of molecularly confirmed Stargardt’s patients revealed a novel frameshift ABCA4 mutation, which accounts for a severly affected phenotype with a relatively early onset of the disease and a rapid deterioration in visual acuity. Conversely, a novel missense mutation was found in one of our mildly affected patients with a later onset of the disease and a significantly better visual acuity. Our results are consistent with previous findings, according to which truncating and severely misfolding mutations are associated with an early-onset disease characterized by a primary photoreceptor loss, whereas in patients with milder mutations, photoreceptors are at least initially spared.10
This small case series of STGD patients demonstrates novel genotype/phenotype correlations, which may be useful to counselling of patients. This information may also prove useful in selection of candidates for clinical trials in ABCA4 disease.

References
Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997; 15: 236–246.
Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR . Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. IOVS 2001; 42: 2757–2761.
Webster A, Héon E, Lotery AJ, Vandenburgh K, Casavant TL, Oh KT et al. An analysis of allelic variation in the ABCA4 gene. IOVS 2001; 42: 1179–1189.
Maugeri A, van Driel MA, van de Pol DJ, Klevering BJ, van Haren FJ, Tijmes N et al. The 2588 G—>C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 1999; 64: 1024–1035.
Jaakson K, Zernant J, Külm M, Hutchinson A, Tonisson N, Glavac D et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003; 22: 395–403.
Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB . The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene Graefes. Arch Clin Exp Ophthalmol 2005; 243: 90–100.
Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res 2005; 37: 159–167.
Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet 1999; 64: 422–434.
Zernant J, Schubert C, Im KM, Burke T, Brown CM, Fishman GA et al. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci 2011; 52 (11): 8479–8487.
Shroyer NF, Lewis RA, Allikmets R, Singh N, Dean M, Leppert M et al. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res 1999; 39: 2537–2544.
Acknowledgements
We thank ‘Fight against Blindness’ as the funder of the above project. We are also grateful to the Wessex Regional Genetics Laboratory (Dr David Robinson) for DNA extraction and sample processing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work was previously presented in ARVO, 6–10 May 2012.
Rights and permissions
About this article
Cite this article
Gemenetzi, M., Lotery, A. Phenotype/genotype correlation in a case series of Stargardt’s patients identifies novel mutations in the ABCA4 gene. Eye 27, 1316–1319 (2013). https://doi.org/10.1038/eye.2013.176
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2013.176
