
Abstract
Purpose
Proliferative vitreoretinopathy (PVR) is the leading cause of failure of surgery for rhegmatogenous retinal detachment. Although indirect evidence suggests that this disease might be autoimmune in nature, direct proof for this hypothesis is lacking. The purpose of this study was to determine in a murine model whether PVR can develop in the absence of T- or B-cell immunity.
Methods
Four- to six-week-old Rag-1 gene knockout (KO) and congenic wild-type mice (WT) on the C57.Bl/6 background were studied. PVR was induced by intravitreal injection of 3 μl dispase at the concentration of 0.2 U/μl. PVR development was monitored by electroretinograms, the macroscopic observation of hemorrhage, cataract, retinal folds, and of an uneven iris, as well as the histological detection of epiretinal membranes on haematoxylin–eosin stained tissue. Additionally, immunofluorescence analysis was performed. These manifestations of PVR were assessed 1, 2, 4, 6, and 8 weeks after the intravitreal injection.
Results
The data showed that the immune-deficient Rag-1 KO mice developed PVR with similar kinetics and severity as did the fully immune competent congenic WT mice. Carboxyfluorescein diacetate succinimidyl ester-labeled T cells that are specific for ovalbumin were detected in the inflamed vitreous and retina showing that T cells that are not specific for autoantigens present in the eye can migrate to PVR lesions. Therefore, the mere presence of T cells in PVR lesions does not imply an autoimmune pathogenesis.
Conclusion
This study suggests that T- and B-cell immunity is not essential for the induction of PVR.
Similar content being viewed by others


Introduction
Proliferative vitreoretinopathy (PVR) can occur as a severe complication after surgery for rhegmatogenous retinal detachment.1 It is characterized by the formation of a scar-like fibrous tissue in the vitreous that contains myofibroblasts derived form transdifferentiated retinal pigment epithelial cells or glial cells.2, 3 The pathogenesis of PVR is still not completely understood. Indirect evidence—but no proof—has been provided in favor of an autoimmune hypothesis as the consequence of which PVR frequently has been referred to as autoimmune complication of eye surgery. The autoimmune hypothesis has been apparently supported by the observation that a PVR-like disease can be induced in rabbits by immunization with the retinal autoantigens opsin, S antigen (S-Ag), and interphotoreceptor retinoid-binding protein.4 In addition, apparently arguing for the autoimmune hypothesis, PVR patients display signs of active immune processes in their epiretinal or subretinal membranes, vitreous cavity, subretinal fluid, and serum samples.5, 6, 7, 8, 9, 10, 11 Thus, deposits of IgG, IgA, and IgE antibodies and complement fragments C1q, C3c, and C3d can be detected in the epiretinal or subretinal membranes of PVR lesions, along with infiltrating CD4 and CD8 T cells, B cells, and macrophages. Moreover, increased expression of CD95-ligand and/or HLA-DR molecules on these immune cells suggests that they are in an activated state. Similar signs of immune activity have been reported for the cells present in the vitreous cavity or subretinal fluid of PVR patients.8, 12, 13, 14, 15 Sera of PVR patients contain increased concentrations of S-Ag16 and S-Ag-specific autoantibodies.17 The presence of these autoantibodies in particular has been thought to support the autoimmune hypothesis because the involvement of antigen-specific CD4 cells, so called T-cell help, is a prerequisite for B-cell responses to mammalian protein antigens in general, and to protein antigens of the eye in particular. Therefore, the presence of autoantibodies directed against protein antigens of the eye implies that auto reactive CD4 cells with the same protein antigen specificity have engaged in an autoimmune response.18 Although all the above findings are consistent with an autoimmune hypothesis for PVR, they do not prove it, and it has been difficult to establish whether the immune reactions seen represent the cause of the disease, or merely accompany it.1
Unambiguous proof for or against the autoimmune hypothesis of PVR requires a model in which the inflammatory sequelae after ocular injury can be studied in the presence or absence of a functional immune system. The classic PVR model does not readily lend itself to such a study because there are no congenic immunodeficient/immunesufficient rabbit strains. We therefore established and studied a murine PVR model that involved C57.Bl/6 wild-type (WT) mice (that have a fully functional immune system) and congenic Rag-1 gene knockout (KO) mice (that are profoundly immunodeficient as they lack the gene that is required to generate antigen receptors for T and B lymphocytes). The purpose of this study was to determine whether T and B cells capable of mounting an autoimmune response are required for the development of PVR.
Materials and methods
Mice
B6. 129S7-Rag1tmiMom/J mice,19 4–6 week old, on a C57.Bl/6 background, were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and used to set up a breeding colony. Homozygous C57.Bl/6-Tg (OT-II.2a)-Rag1 tm1Mom mice,20 bearing transgenes for the expression of an ovalbumin (OVA) -specific T-cell receptor (TCR), were purchased form Taconic Farms, Inc. (Germantown, NY, USA). These mice were bred at our facility to the Rag–/– background to assure exclusive T-cell expression of the TCR transgenes. Animal husbandry and experimental procedures were approved by the Animal Research Committee of Zhongshan Ophthalmic Center, Sun Yat-sen University. All animals were housed in a specific pathogen-free biohazard level-2 facility maintained by the Zhongshan Ophthalmic Center, Sun Yat-sen University (Guangzhou, China) in accordance with the ‘Association of Assessment and Accreditation of Laboratory Animal Care’ (AAALAC) guidelines.
Murine PVR model induced by intravitreal dispase injection
Murine PVR was induced by dispase (GIBCO, Tokyo, Japan) as previously described.21 Four to six weeks old female mice were anesthetized with 4.3% chloral hydrate (0.01 ml/g, from Zhongshan Ophthalmic Center, Sun Yat-sen University). Pupils were dilated with 0.5% tropicamide (from Shenyang SinQi Pharmaceutical Co., Ltd, Shenyang, China). Intravitreal injections were made in the dorsonasal quadrant of the right eye. Three μl dispase at the concentration of 0.2 U/μl was injected into the vitreous cavity with a Hamilton syringe fitted with a 30-G needle. Control animals were injected with 3 μl of sterile saline. All experimental procedures adhered to guidelines of the ‘Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Ophthalmic and Vision Research’.
Follow-up examinations, PVR assessment
After intravitreal injection, at the time points specified, the eyes were examined with a surgical microscope. Corneal- and lens opacities, intravitreal hemorrhage, and the fundus were assessed at each time point. Intravitreal hemorrhage and cataract development are the hallmarks of PVR, but might not be stringent enough criteria for its unambiguous diagnosis in particular after mechanical injury of the eye.22 Therefore, for this study, we adopted an evaluation system introduced by Cantó Soler et al21 that in addition to (a) hemorrhage and (b) cataract requires the development of (c) retinal folds, of (d) epiretinal membranes (EM), and (e) of an uneven iris (the latter indicating retinal traction detachment). Moreover, all these hallmarks of the disease were recorded at 1, 2, 4, 6, and 8 weeks after the injection of the eye. To quantify the severity of the ocular lesions, the cumulative score for the PVR symptoms a–e was established over the entire observation period.
Electroretinography (ERG)
Electroretinograms were obtained from mice at 1, 2, 4, 6, and 8 weeks after intravitreal injection in a full-field dome using methods that are analogs to those commonly used for humans in ophthalmology. The stimuli used were according to the specifications by the ‘International Society for Clinical Electrophysiology of Vision’ standard.23 The eyes were dilated with 0.5% tropicamide and dark-adapted for at least 30 min. Mice were anesthetized with a saline solution containing ketamine hydrochloride (30 mg/kg) and chlorpromazine hydrochloride (15 mg/kg). A gold-wire coil placed on one cornea was referenced to a needle electrode in the scalp. A needle electrode in the tail served as ground. The recordings were made using the Roland Ganzfeld system and PC-based signal acquisition and analysis software (Fa. Roland Consult, Brandenburg, Germany). The contralateral untreated eyes were used as controls.
Tissue preparation, histological, and immunofluorescence analysis
For histochemical studies, mice were killed at the post-injection time points specified, eyes dissected, and cryopreserved using OCT (Optimal Cutting Temperature, Sakura Finetek USA, Inc., Torrance, CA, USA). For immunofluorescence analysis and haematoxylin–eosin (H&E) staining, consecutive 6 μm thick sections of each sample were cut and thaw-mounted onto poly-L-lysine-coated glass slides. For confocal microscopy, double immunostaining was done using two primary antibodies (incubating for 20 h at room temperature with both, simultaneously) and then incubating with the secondary antibodies for 1 h in the dark. The following marker-specific antibodies were used to detect cell types: astroglial cells (glial acidic fibrillary protein, GFAP, mouse monoclonal specific for mouse GFAP, used at 1 : 500, from CHEMICON International, Inc., Temecula, CA, USA), Müller cells (glutamine synthase (GS), rabbit polyclonal specific for mouse GS, 1 : 50, from Abcam, Cambridge, UK), RPE cells (RPE65, mouse monoclonal specific for mouse RPE, 1 : 100, from Abcam), fibroblasts (alpha smooth muscle actin, α-SMA, rabbit polyclonal specific for mouse α-SMA, 1 : 100, from Abcam), macrophages (F4/80, rat monoclonal specific for mouse F4/80, 1 : 10, from Abcam), T lymphocytes (anti-CD3, rat monoclonal specific for mouse CD3, 1 : 50, from R&D Systems, Inc., Minneapolis, MN, USA), and NK cells (anti-CD56, rabbit polyclonal antibody specific for mouse CD56, 1 : 100 from Boster Biological Technology, WuHan, China). Three secondary antibodies were used in this study: R-phycoerythrin-conjugated goat anti-rat IgG (1 : 10, from Southern Biotechnology Associates, Inc., Birmingham, AL, USA), R-phycoerythrin-conjugated goat anti-mouse IgG (1 : 10, from Southern Biotechnology Associates, Inc.) and fluorescein isothiocyanate-labeled goat anti-rabbit IgG (1 : 10, from KPL, Gaithesburg, MD, USA). Sections were washed four times with PBS (5 min each time) and mounted on coverslips in Antifade Solution (Applygen Technologies Inc., Beijing, China) for observation with a fluorescence microscope.
Tracking adoptively transferred OT-II cells in mice
OT-II mice are transgenic for a pair of TCR chains that encode a TCR specific for OVA peptide 323–339 when presented on the MHC class II molecule, I-Ab. Therefore, all CD4 cells are OVA 323–339-specific in OT-II mice that are bred with Rag KO mice to avoid the rearrangement of endogenous TCR genes. Such OVA 323–339 peptide-specific TCR transgenic CD4+ T cells were isolated from spleens and lymph nodes of OT-II Rag KO mice. CD4+ T cells, adjusted to 4 × 106 cells/ml, and were activated by culturing with the OVA 323–339 peptide (1 μg/ml, from Anaspec, Inc., San Jose, CA, USA) for 5 days in a humidified incubator at 37 °C, 9% CO2. Then the OVA-specific CD4+ T cells were washed and incubated with Carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Inc., Grand Island, NY, USA) at a final concentration of 4 μg/ml at 37 °C for 10 min. Then the cells were washed, adjusted to 4 × 106 cells/ml, and one million cells (in 0.25 ml) were injected into the mice via their lateral tail vein. The recipient Rag KO mice were at the same time treated with intravitreal dispase injection and were killed at five time points (after 12, 24, 48, 72, and 96 h). Eyes and spleens were processed for OCT. Frozen sections were stained with anti-CD3 antibody (the second antibody was R-phycoerythrin-conjugated goat anti-rat IgG) and the number of CFSE, CD3 double positive cells was established. Three mice per time point were studied, and experiments for each condition were reproduced at least twice.
Statistical analysis
PVR incidences were compared among the time points using χ2 test. Data of Skot ERG were compared by Student's t-test. A value of P<0.05 was considered significant.
Results
Rag-1 KO mice develop PVR
Figure 1 shows that immune-deficient Rag-1 KO mice develop all cardinal features of PVR after intravitreal injection of dispase (Figure 1a). Formation of marked EMs and tractional retinal detachment was seen in the vitreous cavity (Figure 1b). In the EM cells, characteristic of PVR were detected, namely glial cells, fibroblasts, RPE-, and Mûller cells staining for GFAP, α-SMA, RPE65, and GS, respectively (Figure 1c).

Rag-1 KO mice develop all cardinal features of PVR. Representative PVR manifestations are shown for a total of 45 Rag-1 KO mice studied 2–8 weeks after injection of dispase. (a) Traction membrane in dispase-injected eye as indicated by arrow. (b) Proliferative membrane in vitreal cavity and retinal detachment as indicated by arrow. (c) Immunofluorescence staining for α-SMA (c-1), GFAP (c-2), GS (c-5), and RPE-65 (c-6) in the epiretinal membrane (EM), and Hoechst 33342 staining to mark cell nuclei with the EM (c-3 and c-7). c-4 is the merged picture of c-1–c-3, and c-8 is the merged picture of c-5–c-7 (triple staining).
Rag-1 KO and WT mice show similar kinetics and severity of PVR disease
The rate at which PVR developed after dispase injection was monitored in Rag-1 KO and WT mouse strains to assess the severity and kinetics of PVR development in the absence or presence of a functional adaptive immune system. In both type of mice, the macroscopic signs of the disease developed in parallel. In both the groups, the first signs of PVR appeared 2 weeks after dispase injection, affecting about 25% of the mice (Figure 2a). The percentages of PVR-positive mice gradually increased in the subsequent weeks reaching about 70% for both the groups by 8th week. Statistical analysis showed no significant difference between the Rag KO and the WT group at weeks 2, 4, 6, and 8 with P-values of 0.2163, 0.2503, 0.2176, and 0.2642, respectively.

Development of clinical PVR in Rag-1 KO and WT mice after dispase injection. A total of 10 mice in each group were injected intraocularly with 0.2 U/μl dispase. At the time points specified, the eyes were examined for: (a) macroscopic presentation of PVR symptoms; and (b) histological evidence for PVR as established on H&E-stained sections. (c) Skot ERGs recording α- and β-wave amplitudes. The contralateral untreated eyes were used as controls. The data in both panels represent the cumulative results obtained on 25 Rag-1 KO mice after dispase injection.
Histological studies were performed to compare the kinetics and severity of the dispase-induced disease in KO and WT mice (Figure 2b). Mice of both groups were killed at 1, 2, 4, 6, and 8 weeks and HE-stained frozen sections of their eyes were studied. PVR kinetics and severity did not markedly differ between the Rag KO and the WT mice at weeks 2, 4, 6, and 8.
The severity of PVR was also compared in the two groups of mice by Skot ERG (Figure 2). For dispase-injected eyes there was no statistically significant difference in α-wave (P=0.3454) and β-wave amplitudes (P=0.5374) of the WT and KO group. The contralateral untreated eyes were used as controls. The untreated eyes of dispase-injected mice also showed no significant difference in α-wave (P=0.4053) or β-wave amplitudes (P=0.5039).
In WT and Rag-1 KO mice neutrophilic infiltration dominates the initial inflammatory reaction, occurring with similar kinetics
We harvested eyes from KO and WT mice that were untreated, or 4, 8, 12, 24, and 48 h and 5 days after injection of 3 μl of dispase. The eyes of control mice were injected with 3 μl saline, and studied at these time points. HE staining showed that polymorphonuclear neutrophils were the main infiltrating cell type in the eyes of dispase-injected KO and WT mice. In both strains, the neutrophils started to appear 8 h post injection, peaked at 24–48 h, and decreased by day 5 (Figure 3). In contrast, eyes of the saline-injected KO or WT mice did not show an infiltrate at any of these time points.

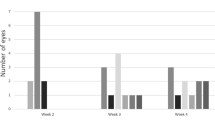
Early inflammatory infiltration occurred at the same speed in WT and KO mice. (a) H&E staining of the retina after dispase or saline injection. (b) H&E staining of the aqueous angle. Arrows indicate neutrophils. (c) Immunofluorescence analysis showing the absence of T cells (CD3+), macrophages (F4/80+), and NK cells (CD56+) 48 h after dispase injection.
Bystander T cells are recruited into the PVR infiltrate
We detected T cells in PVR lesions of WT mice at the chronic stage of the inflammatory process. Our observation, however, that PVR develops at a similar rate and severity in Rag-1 KO and WT mice suggests that these T cells in PVR regions might be bystander cells, that is, T cells with specificity for irrelevant antigens that are non-specifically recruited to the site of inflammation. To test this possibility, we studied whether T cells specific for OVA (an irrelevant antigen in the context of PVR) would migrate into PVR lesions. OVA-specific TCR transgenic T cells (that in addition were Rag-1−/− to assure that they express no additional endogenous TCR chains, and thus are specific for OVA only) were used to test this hypothesis. Such T cells were labeled with CFSE and injected into dispase-treated mice. CFSE-labeled cells were detected in the inflamed vitreous and retina at 48 and 72 h time point (Figure 4) showing that bystander T cells can migrate to PVR lesions. Thus, the presence of T cells in the PVR infiltrate does not imply that those T cells are specific for autoantigens expressed in the eye; their presence does not imply an autoimmune pathogenesis.

Detection of OVA-specific T cells in PVR lesions at 48 and 72 h time point. OVA-specific, OT II TCR-transgenic T cells were labeled with CFSE and injected i.v. into congenic Rag-1 KO mice that received a simultaneous intraocular dispase injection. The mice were killed 12, 24, 48, 72, and 96 h later and cryosections of their eyes were studied for the presence of green-fluorescent (CFSE positive) cells. Hoechst 33342 dye was used to stain nuclei of the tissue blue. Anti-CD3 staining is show in red.
Discussion
The data presented here establish that autoimmunity is not required for the development of dispase-induced PVR in C57.Bl/6 mice as T- and B-cell-deficient Rag-1 KO animals develop PVR with similar kinetics and severity than WT mice. We further show that the presence of T cells in PVR lesions does not provide evidence for an autoimmune etiology, as T cells with irrelevant specificity for autoantigens present in the eye (that is, T cells specific for OVA) are readily recruited to PVR lesions. To our knowledge this is the first report that clearly establishes that autoimmune reactions are not required for the pathogenesis of PVR. As detailed in the Introduction, the autoimmune hypothesis has relied on indirect evidence, such as the presence of T cells, which, however, can also be interpreted within the framework of non-specific inflammatory reactions mediated by the cells of innate immunity. Data that were interpreted to argue against the autoimmune hypothesis are also rather indirect. Thus, Bali et al24 showed that interleukin-10 had no effect on the clinical progression of PVR. Although IL-10 can inhibit T cells, it promotes B-cell responses25 and as such may have little net impact on an autoimmune disease.
In contrast to the findings of this study it has been reported that S-Ag-specific autoantibodies can be detected in PVR.16 The presence of such antibodies implies that S-Ag-specific autoreactive B cells have engaged in an autoimmune response in PVR, and as the production of such antibodies is dependent on antigen-specific help by CD4 cells, by interference, that is, an autoimmune T-cell response has been engaged. Autoimmune reactions, however, can accompany tissue injury without causing immune pathology.26 The S-Ag-specific autoantibodies that accompany PVR might be indicative of such non-pathogenic, secondary, autoimmune reactions. What then makes the difference between pathogenic and non-pathogenic autoimmunity? In the case of S-Ag, the answer seems to be that it is the engagement of T cells, rather than of B cells and antibodies. Active immunizations with S-Ag can cause an autoimmune disease, experimental allergic uveitis that entails PVR-like manifestations. While S-Ag-specific antibodies are non-pathogenic, CD4 cells of certain effector class(es) can mediate the autoimmune pathology. The in vivo differentiation of such CD4 cells requires the injection of S-Ag with complete Freund's adjuvant (CFA).27 For a related autoimmune model, experimental allergic encephalomyelitis (that is, mediated by CD4 cells after immunization with an autoantigen of the brain), we recently reported that CFA—unlike other adjuvants such as CpG—causes the differentiation of autoantigen-specific Th17 cells, and that this IL-17 is required for the development of the autoimmune disease.28 Thus, the CD4 cells that provide autoantigen-specific help towards the production of eye-antigen-specific autoantibodies might not cause autoimmune pathology unless they are of the Th17 lineage.
Our data show that dispase is a most potent activator of the innate immune system. In Rag 1 KO mice, in the absence of amplification by T- and B-cell immunity, dispase triggers an inflammatory reaction strong enough to result in full-blown PVR. This severe non-specific inflammation mediated by cells of innate immunity seems to suffice to bring about the changes that had been interpreted as evidence for the autoimmune hypothesis: deposits of antibodies and of complement, and the presence of CD4 and CD8 T cells in the eye. Along these lines, it should be noted that the fact that these T cells display an activated phenotype in PVR lesions does not imply that they actively recognize antigen at the site. While resting T cells home to lymph nodes, activated T cells preferentially migrate to sites of inflammation. Tissue injury leads to non-specific inflammation. As an intrinsic part the non-specific inflammatory reaction, white blood cells of all lineages are recruited to the injured site. Neutrophils dominate the infiltrate being the prevalent cell type in blood, but T cells are also recruited.29 In brief, tissue injury triggers the local release of prostaglandins and vasoactive amines that cause local vasodilatation. As the capillary bed widens, blood flow slows down, and cells in the blood stream get to contact the endothelial cells. Under the influence of IL-1 and TNFα produced by macrophages in the inflamed tissue, the endothelial cells in the local post capillary venules start to express cell adhesion molecules, including selectins. Expressing ligands for selectins, leukocytes attach to these endothelia, and under the guidance of PECAM proteins, the leukocytes migrate through the vessel wall. Once in the interstitial space, the leukocytes follow the chemotactic gradient to continue their migration to the actual site of the injury. As neutrophils prevail in the blood stream, in the first 12 h of inflammation polymorphonuclear cells dominate the infiltrate. These cells are short lived, however, and during sustained non-specific inflammation long-lived leukocytes such as macrophages and lymphocytes can become dominant. The OVA-reactive T cells fall in this latter category being recruited to the site of inflammation irrespective of their antigen specificity.
Our data do not disprove the autoimmune hypothesis for humans, or that in murine models secondary autoimmune reactions could emerge possibly further aggravating PVR. It, however, shows that the full clinical and histological presentation of PVR can occur in the complete absence of specific T- or B-cell immunity. Moreover, our data show that already the initial non-specific inflammatory reactions mediated by the innate immune system suffice to trigger these clinical manifestations before the amplification by adaptive immune responses emerge.
PVR models have also been generated in the monkey30 and rabbit,31 and those models have been preferably studied because the larger eyes of those animals facilitate the clinical follow up. The murine model, however, is unique in as much that inbred strains are available for avoidance of genetic variability within the model. Moreover, our data show that the initial non-specific inflammatory reactions mediated by the innate immune system is already sufficient to trigger these manifestations. We believe that of the PVR models available for different species, dispase-induced PVR in C57.Bl/6 mice will gain prevalence because the abundance of knockout/knock in mice available facilitates mechanism-oriented PVR research. Our use of the Rag-1 KO mice for PVR research is the first step in that direction.
A better understanding of the pathogenic mechanisms that underlie PVR will help improve the therapeutic approach to the disease. While it is unclear how well murine dispase-induced PVR models the human disease, our data clearly show that the innate immune system alone can have a major role in inflammatory eye pathology. In spite of the prevalence of the autoimmune hypothesis, in the absence of an alternative that would permit to selectively inactivate autoreactive T cells, the present treatment of human PVR in the clinic targets the innate immune system. Triamcinolone acetonide is being used as an anti-inflammatory drug for the early stage treatment and anti-proliferation treatment for the advanced stage. If dispase-induced murine PVR indeed models the human disease then future efforts to refine an anti-inflammatory approach that targets innate immunity is likely to improve the present therapeutic success while approaches that inhibit T cells might not be successful.

References
Pastor JC . Proliferative vitreoretinopathy: an overview. Surv Ophthalmol 1998; 43: 3–18.
Pastor JC, de la Rua ER, Martin F . Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res 2002; 21: 127–144.
Agrawal RN, He S, Spee C, Cui JZ, Ryan SJ, Hinton DR . In vivo models of proliferative vitreoretinopathy. Nat Protoc 2007; 2: 67–77.
Broekhuyse RM, Rademakes AJ, Van Vugt AH, Winkens HJ . Autoimmune responsiveness to retinal IRBP, S-antigen and opsin in proliferative vitreoretinopathy. Exp Eye Res 1990; 50: 197–202.
Baudouin C, Fredj-Reygrobellet D, Gordon WC, Baudouin F, Peyman G, Lapalus P et al. Immunohistologic study of epiretinal membranes in proliferative vitreoretinopathy. Am J Ophthalmol 1990; 110: 593–598.
Charteris DG, Hiscott P, Grierson I, Lightman SL . Proliferative vitreoretinopathy. Lymphocytes in epiretinal membranes. Ophthalmology 1992; 99: 1364–1367.
Charteris DG, Hiscott P, Robey HL, Gregor ZJ, Lightman SL, Grierson I . Inflammatory cells in proliferative vitreoretinopathy subretinal membranes. Ophthalmology 1993; 100: 43–46.
Tang S, Scheiffarth OF, Wildner G, Thurau SR, ELund O . Lymphocytes, macrophages and HLA-DR expression in vitreal and epiretinal membranes of proliferative vitreoretinopathy. An immunohistochemical study. Ger J Ophthalmol 1992; 1 (3-4): 176–179.
Liversidge J, Sewell HF, Forrester JV . Interactions between lymphocytes and cells of the blood-retina barrier: mechanisms of T lymphocyte adhesion to human retinal capillary endothelial cells and retinal pigment epithelial cells in vitro. Immunology 1990; 71: 390–396.
Proenca R, Carvalho M, Proenca D, Verissimo J, Regadas I, Travassos A . HLA antigens and lymphocytes in proliferative vitreoretinopathy. Graefes Arch Clin Exp Ophthalmol 1994; 232 (1): 25–32.
Weller M, Heimann K, Bartz-Schmidt KU, Fontana A, Esser P . CD 95 expression in traumatic proliferative vitreoretinopathy: a target for the induction of apoptosis. Ger J Ophthalmol 1995; 5: 332–337.
Baudouin C, Brignole F, Bayle J, Fredj-Reygrobellet D, Lapalus P, Gastaudet P . Class II histocompatibility antigen expression by cellular components of vitreous and subretinal fluid in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 1991; 32: 2065–2072.
El-Ghrably IA, Dua HS, Orr GM, Fischer D, Tighe PJ . Intravitreal invading cells contribute to vitreal cytokine milieu in proliferative vitreoretinopathy. Br J Ophthalmol 2001; 85: 461–470.
Martin F, Pastor JC, De La Rua ER, Mayo-Iscar A, García-Arumí J, Martínez V et al. Proliferative vitreoretinopathy: cytologic findings in vitreous samples. Ophthalmic Res 2003; 35: 232–238.
Canataroglu H, Varinli I, Ozcan AA, Canataroglu A, Doran F, Varinli S . Interleukin (IL)-6, interleukin (IL)-8 levels and cellular composition of the vitreous humor in proliferative diabetic retinopathy, proliferative vitreoretinopathy, and traumatic proliferative vitreoretinopathy. Ocul Immunol Inflamm 2005; 13: 375–381.
Grisanti S, Heimann K, Wiedemann P . Immune response to specific molecules of the retina in proliferative vitreoretinal disorders. Graefes Arch Clin Exp Ophthalmol 1994; 232: 302–307.
Koerner F, Merz A, Gloor B, Wagner E . Postoperative retinal fibrosis--a controlled clinical study of systemic steroid therapy. Graefes Arch Clin Exp Ophthalmol 1982; 219: 268–271.
Beissert S, Schwarz A, Schwarz T . Regulatory T cells. Invest Dermatol 2006; 126: 15–24.
Mombarets P, Lacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE . RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992; 68: 869–877.
Barnden MJ, Allison J, Heath WR, Carbone FR . Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 1998; 76: 34–40.
Cantó Soler MV, Gallo JE, Dodds RA, Suburo AM . A mouse model of proliferative vitreoretinopathy induced by dispase. Exp Eye Res 2002; 75: 491–504.
Kralinger MT, Kieselbach GF, Voigt M, Hayden B, Hernandez E, Fernandez V et al. Experimental model for proliferative vitreoretinopathy by intravitreal dispase: limited by zonulolysis and cataract. Ophthalmologica 2006; 220: 211–216.
Marmor MF, Holder GE, Seeliger MW, Yamamoto S . International Society for Clinical Electrophysiology of Vision. Standard for clinical electroretinography (2004 update). Doc Ophthalmol 2004; 108: 107–114.
Bali E, Willermain F, Caspers-Velu L, Dubois C, Dehou MF, Velu T et al. IL-10 in vivo gene expression in a cell-induced animal model of proliferative vitreoretinopathy. Int J Mol Med 2003; 12: 305–310.
Qian S, Li W, Li Y, Fu F, Lu L, Fung JJ et al. Systemic administration of cellular interleukin-10 can exacerbate cardiac allograft rejection in mice. Transplantation 1996; 62: 1709–1714.
Sontheimer RD . Subacute cutaneous lupus erythematosus: 25-year evolution of a prototypic subset (subphenotype) of lupus erythematosus defined by characteristic cutaneous, pathological, immunological, and genetic findings. Autoimmun Rev 2005; 4: 253–263.
Uchio E, Kijima M, Ishioka M, Tanaka S, Ohno S . Suppression of actively induced experimental autoimmune uveoretinitis by CD4+ T cells. Graefes Arch Clin Exp Ophthalmol 1997; 235: 97–102.
Hofstetter HH, Kovalovsky A, Shive CL, Lehmann PV, Forsthuber TG . Neonatal induction of myelin-specific Th1/Th17 immunity does not result in experimental autoimmune encephalomyelitis and can protect against the disease in adulthood. J Neuroimmunol 2007; 187: 20–30.
Sorokin L . The impact of the extra cellar matrix on inflammation. Nat Rev Immunol 2010; 10: 712–723.
Machemer R, Norton EW . Experimental retinal detachment in the owl monkey: methods of production and clinical picture. Am J Ophthalmol 1968; 66: 388–395.
Frenzel EM, Neely KA, Walsh AW, Cameron JD, Gregerson DS . A new model of proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 1998; 39: 2157–2164.
Acknowledgements
This study was supported by the Natural National Science Foundation of China (30973258) and the Science and Technology Foundation of Guang Dong Province of China (2010A090200074).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhang, W., Tan, J., Liu, Y. et al. Assessment of the innate and adaptive immune system in proliferative vitreoretinopathy. Eye 26, 872–881 (2012). https://doi.org/10.1038/eye.2012.52
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.52
Keywords
This article is cited by
-
Antibody-Drug Conjugate (ADC) Research in Ophthalmology—a Review
Pharmaceutical Research (2015)
-
Factor Xa and thrombin stimulate proinflammatory and profibrotic mediator production by retinal pigment epithelial cells: a role in vitreoretinal disorders?
Graefe's Archive for Clinical and Experimental Ophthalmology (2013)
