
Abstract
Purpose
(1) To evaluate the spectrum of BEST1mutations within Australian Best Disease or vitelliform macular dystrophy (VMD) pedigrees, including any novel mutations; (2) to analyse the range of clinical presentations of this cohort; (3) to determine any possible genotype–phenotype correlations and (4) to compare clinical data of patients with phenotypic VMD, both with and without a BEST1mutation.
Patients and methods
Patients with suspected VMD were referred to clinical centres for ophthalmological assessment and genetic screening. When a mutation was identified in a proband, further family members were invited for clinical and genetic screening.
Results
We identified 42 patients with one of 13 BEST1mutations. Seven mutations were novel. There were a further 14 probands in whom a BEST1mutation was not identified. Median visual acuity in both VMD (mutation positive) and clinical VMD (no BEST1mutation identified) groups reached driving standards (6/12 or better).
Conclusion
We did not identify any firm genotype–phenotype correlations in our Australian VMD pedigrees, in which there was a spectrum of BEST1mutations and marked variation in clinical presentation. Genetic screening remains the gold standard for VMD diagnosis. Patients should be counselled that visual acuity might remain at or above driving standards in at least one eye even in the presence of a BEST1mutation.
Similar content being viewed by others


Main
Vitelliform macular dystrophy (VMD), first described by German ophthalmologist Friedrich Best in 1905 (reviewed in Best1), is an autosomal dominantly inherited eye disease, with variable penetrance and expressivity (OMIM 153700).2, 3, 4 Most cases are due to mutations in the bestrophin 1 gene (BEST1; formerly named VMD2) located on chromosome 11q12-q13. Although the precise role of the encoded protein is still debated, bestrophin 1 is thought to belong to a family of calcium-activated chloride channels,5, 6, 7, 8 and has been identified on the basolateral plasma membrane of retinal pigment epithelium (RPE).9, 10, 11, 12 It is thought to be a separate disease entity to adult vitelliform macular dystrophy (AVMD), which is a rare disorder, with autosomal dominant inheritance, and variable penetrance and expressivity. Mutations in both the RDS and BEST1 genes are responsible for AVMD, and it is considered as a subset of pattern dystrophies. Unlike VMD, AVMD characteristically manifests from third to fifth decades.13, 14, 15
Since its identification, over 120 disease-causing mutations have been described in the BEST1 gene.16 The majority of mutations are missense and located in the N-terminal of the gene. Marked pleiotropy has been observed with mutations in the BEST1 gene, with clinical manifestations, including autosomal recessive bestrophinopathy, autosomal dominant vitreoretinal choroidopathy (ADVIRC OMIM 193220),17 AVMD (OMIM 608161),13, 18 and recessive and dominant forms of retinitis pigmentosa,19 as well as dominantly inherited MRCS (comprising microcornea, rod-cone dystrophy, early-onset cataract, posterior staphyloma).17, 20, 21, 22 Nonetheless, VMD is the most common disease associated with BEST1 mutations.
Clinically, VMD is characterised by progressive visual loss of variable onset. There have been two peaks of disease onset described: one in early childhood and the other occurring in late adolescence or early adulthood. However, VMD has known disease phenotypic variability and onset may be as late as the fifth decade.2, 23, 24, 25, 26 Typically, patients present with blurred vision, loss of central acuity or metamorphopsia.24 The electrophysiological hallmark of VMD is a reduced Arden index on electro-oculogram (EOG).27, 28 In patients with VMD, the light peak-dark trough ratio is usually <1.5 in the presence of a normal electro-retinogram (ERG).29, 30 In the absence of genetic testing, the carrier state may often only be identified in VMD by an abnormal EOG;29 however, some studies have shown the preservation of a normal EOG despite the presence of a BEST1 mutation.2, 16, 25, 31
Considerable variation in VMD disease expressivity has been observed.16, 26, 32, 33, 34, 35 The funduscopic appearance of VMD varies depending on disease severity and stage. Traditionally, five main patterns of retinal dystrophy are described:28 (1) previtelliform (normal macula or only slight RPE disturbance); (2) vitelliform (an ‘egg yolk’ lesion); (3) pseudohypopyon (sinking of the yellow deposits inferiorly); (4) vitelliruptive (‘scrambled egg’ appearance with partial resorption of yellow deposits); (5) atrophic. Additionally, the disease may be complicated by the development of a choroidal neovascular membrane. The aim of this study was to describe both the phenotypic and genotypic spectrum of BEST1-related VMD in an Australian cohort. We also sought to investigate specifically the clinical features in patients diagnosed clinically with VMD in whom a BEST1 mutation was not identified.
Materials and methods
Ethical approval
This study was approved by the human research ethics committee of the Royal Victorian Eye and Ear Hospital (RVEEH), Melbourne, Australia, and conformed to the tenets of the Declaration of Helsinki. Informed consent was obtained from all participants following explanation of the nature and purpose of the study along with expected outcomes.
Case identification and subject recruitment
Patients with presumed VMD were identified through the Ocular Diagnostic Clinic of the RVEEH, the Ophthalmology Clinic of the Royal Hobart Hospital and the Eye Hospital Launceston, Tasmania. Other cases were referred from private clinics in Melbourne and Tasmania.
Clinical evaluation and statistical analysis
A clinical diagnosis of VMD in probands was determined primarily by evidence of a characteristic fundus appearance of a VMD-type macula lesion. Reduced visual acuity, abnormal colour vision, a central scotoma on Goldman visual field testing, an Arden index of <1.8 on EOG, and changes on ocular coherence tomography (OCT) were also considered when making the clinical diagnosis. This broad clinical definition was used given the wide variability in presentation of BEST1 mutation carriers, including patients with a BEST1 mutation but normal EOGs. Genealogical pedigrees were constructed where possible to confirm an autosomal dominant mode of inheritance. For all clinical testing, each eye was tested separately, beginning with the right. Best-corrected visual acuity (BCVA) in adults was measured using Snellen or logMAR charts. BCVA in children under 5 years was measured using the Kay Picture Test (Kay Pictures, Tring, UK). Patients were divided into four groups depending on the severity of visual loss. These groups were adapted from the Blue Mountains Eye Study:36 group 1, no visual impairment (BCVA 6/12 or better); group 2, mild visual impairment (BCVA <6/12 to 6/18); group 3, moderate visual impairment (BCVA 6/24 to 6/48); and group 4, severe visual loss (6/60 or worse).
Colour vision testing was undertaken using Ishihara's test for colour blindness (Kanehara Ltd, Tokyo, Japan) consisting of 25 printed pseudo-chromatic colour plates. Each eye was given a score, and patients were divided into three groups according to their score out of 25: group 1, normal colour vision (25 correct colour plates in each eye); group 2, mild red-green colour defect (20–24 correct); and group 3, severe red-green colour deficiency (less than 20 correct).
Patients had visual field testing using a Haag–Streit (Berne, Switzerland) International Goldman visual field instrument using a white light with a target size of III. Central scotomas and blind spots, as well as the peripheral visual field, were plotted. Patients were divided into three groups: group 1, no central scotoma; group 2, central scotoma; and group 3, other abnormality (for example, constricted peripheral fields).
EOGs (Medelec Sensor, Old Woking, UK) were performed using either gold or silver/silver chloride discs on the nasal bridge and temples with redux sodium gel (Marker Lab, Slater, IA, USA) for electrode contact. Testing was performed during a light-dark adaptation cycle so that RPE was tested under both scotopic and photopic conditions. The Arden index was calculated for each from the ratio of light peak over the dark trough. An Arden index less than 1.8 was considered abnormal. Patients were divided into three groups: normal (group 1 Arden index >1.8); abnormal (group 2 Arden index 1.8–1.5), and severely abnormal (group 3 Arden index <1.5).
Following pupillary dilation with tropicamide 1%, fundus photography was performed either using a Nidek 3-DX (Nidex, Aichi, Japan) camera or TopCon 50EX camera with ImageNet software (TopCon Medical Systems, Oakland, NJ, USA). OCT was performed using the Stratus OCT (Carl Zeiss Meditec, Inc., Jena, Germany) and central retinal thickness (CRT) for the inner 1 mm zone was measured using the macular scan protocol.37, 38
Comparison of non-parametric continuous variables was performed using the Wilcoxon Mann–Whitney U-test. The χ2 or Fisher's exact test was used in the analysis of categorical variables. Statistical analyses were performed using Intercooled Stata 7.0 for Windows (Stata Corporation, College Station, TX, USA).
Genomic analysis
Samples for DNA extraction were obtained from venipuncture, buccal swab, or saliva using standard techniques. Genomic DNA was extracted using Puregene DNA Purification Kits (Gentra Systems, Minneapolis, MN, USA) or the Oragene DNA extraction kit (DNA Genotek, Ontario, Canada).
Mutation screening was undertaken by the John and Marcia Carver Nonprofit Genetic Testing Laboratory at the University of Iowa, USA (https://www.carverlab.org). The proband was initially screened and when a mutation was identified, cascade screening was performed. Following PCR, exons 2–8 (in which 95% of reported disease-causing variations are located) of BEST1 were directly sequenced. Variant designation was based on the guidelines established by the Human Genome Nomenclature Working Group.39 For all identified variants, cross-species homology was compared between 31 eutherian mammals (data available on request). The predicted effects of the non-synonymous variants were examined using SIFT analysis (http://sift.jcvi.org/) and Polyphen prediction (http://genetics.bwh.harvard.edu/pph). A total of 192 control samples were screened for the BEST1 variations reported in this paper and none were found.
Results
Spectrum of BEST1 mutations
Initial referral of 37 probands with suspected VMD led to recruitment of 135 people within their extended families. After screening all probands, mutations were identified in 15 individuals (40.5%) (Table 1). Following cascade screening, 42 individuals were found to harbour 1 of the 13 mutations in the BEST1 gene. Of the identified mutations, seven (54%) were novel (Table 1). Three families were found to carry the same novel BEST1 mutation (p.Cys221Phe) despite being genealogically unrelated. A total of four non-synonymous (non-disease causing) sequence variations were identified in 14 mutation carriers (Leu37Leu TTA>CTA; IVS4–24 C>T; IVS4 +280 A>T; IVS4 +133 G>C; Ile73Ile ATC>ATA). There were 14 people with clinical VMD in whom mutation screening failed to discover a BEST1 gene mutation. These patients will be referred to as clinical VMD patients.
Clinical spectrum
Of the 42 people found to carry a BEST1 mutation, visual acuity was available for 39 of them. BCVA in the better eye was used for analysis. The mean age of mutation carriers at the time of last clinical review was 43 years. Approximately 46% of mutation carriers were women.
In BEST1 mutation carriers, the BCVA in the better eye ranged from 6/4 to count fingers (CF) (median 6/12). In clinical VMD patients, the BCVA ranged from 6/5 to CF (median 6/12). Table 2 displays the breakdown of age at examination, categorised for BCVA of the patients in their better eye.
Patients were separated into three groups (see Materials and methods) according to their colour vision deficit. Colour vision results were available for 30 (72%) mutation carriers and 12 (85%) clinical VMD patients. A total of 12 mutation carriers (28%) were unable to perform the test because of severe visual loss, being too young or for some other reason. Total 15 (35%) mutation carriers were in group 1 (all Ishihara colour plates correct in either eye), 9 (21%) were in group 2 (between 20 and 24 plates correct), and 6 (14%) were in group 3 (less than 20 correct). Five clinical VMD patients were in group 1 (35%), five in group 2 (35%), and two in group 3 (14%).
In total, 15 BEST1 mutation carriers (36%) had no visual field performed. Considering results only for the right eye in the mutation-carrier group, 19 (46%) had normal Goldman perimetry, 5 (12%) had a central scotoma, and 2 (6%) had constricted peripheral visual fields. In the clinical VMD group, only one patient did not have a visual field performed (7%). Normal perimetry was present in eight patients (57%); four patients had central scotoma (29%); and one patient with a vitelliform macular lesion had a visual field deficit, not consistent with VMD.
EOG results were separated into three groups (see Materials and methods). In the mutation-carrier group, 17 patients (41%) did not have an EOG performed. Seven (17%) mutation carriers had normal EOGs (group 1); three (7%) had abnormal EOGs (group 2), and 11 (26%) had severely abnormal EOGs (group 3). One (2%) mutation carrier had an EOG that could not be interpreted. Therefore, a total of 14 BEST1 mutation carriers out of 22 tested (53%) had EOGs consistent with VMD. In the non-BEST1 VMD group, eight (57%) patients had an EOG performed; five (36%) were normal (group 1), two (6%) were mildly abnormal (group 2), and one (3%) was severely abnormal (group 3).
All fundus stages of VMD were observed in our BEST1 mutation-carrying cohort. Poorer visual acuity was generally associated with atrophic macula changes. The fundus appearances of a selection of patients with BEST1 mutations who also had BCVAs better than 6/7 in both eyes are displayed in Figure 1. The correlation between fundus appearance and OCT central macular thickness findings, for both BEST1 mutation carriers and clinical VMD patients, is displayed in Figure 2. The clinical VMD group, which had no BEST1 mutation identified, were found to have thinner CRTs compared with BEST1 mutation carriers (P<0.001). However, this maybe selection bias, as all the patients in this group were the pooled probands without mutations and with no other family members screened. Table 3 displays visual acuity and OCT findings in our VMD cohorts.

Examples of fundus appearance in people found to carry BEST1 mutations, who also had BCVAs better than 6/7 in both eyes. (a) Individual Fam32-6, aged 50 years, with p.Arg218His BEST1 mutation; (b) individual Fam05-5, aged 65 years, with Ile310Thr BEST1 mutation; (c) individual Fam11-05, aged 48 years, with Cys221Phe BEST1 mutation.

Distribution of central macular thickness (μm) for different clinical macular appearances. BEST1-mutation carrying (○) and non-mutation carrying VMD (Δ) patients are displayed.
The variable OCT findings observed in our VMD cohort are depicted in Figure 3. Similar to previous OCT studies, earlier stage VMD was associated with accumulation of material in the outer retina, with preservation of vision and later lesions showing central clearing of this material and deterioration of visual acuity. People found to carry BEST1 mutations, eyes with a normal clinical appearance on photos and OCT were more likely to have a corresponding visual acuity better than or equal to 6/12 (Fisher's exact P=0.01).

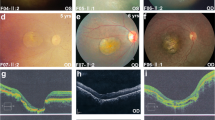
Fundus and optical coherence tomography appearances in BEST1-mutation carrying patients. (a) Individual Fam09-1, aged 16 years, with p.Cys221Phe BEST1 mutation, a vitelliform macular lesion, and a BCVA of 6/7.5; (b) individual Fam05-2, aged 39 years, with p.Ile310Thr. BEST1 mutation and vitelliruptive macular lesion and a BCVA of 6/12; (c) individual Fam11-15, aged 21 years, with p.Cys221Phe BEST1 mutation and atrophic macular lesion, and a BCVA of 6/18.
We had serial photography and clinical information available in a small group of patients. Figure 4 shows serial photos taken at 2 years apart in a VMD patient progressing from vitelliform to vitelliruptive macula lesions with slight worsening of visual acuity.

Natural history of VMD fundus lesions. Images obtained from patient FAM5-1 who carries the p.Ile310Thr BEST1 mutation. Panel (a) taken at age 8 years, with BCVA being 6/30 and 6/24 in his right and left eyes, respectively, and bilateral vitelliform. Panel (b) was taken when the patient was aged 10 years, with BCVA of 6/36 in both the eyes, and bilateral vitelliruptive lesions.
Genotype–phenotype correlations
Patients with confirmed BEST1 mutations in this study had variable disease expression, both within and between families. Interestingly the three families (13 patients; mean age 47 years) with the same mutation in exon 6 (c.662G>T; p.Cys221Phe) had relatively good vision with a median BCVAR 6/12 (range 6/4–6/38) and BCVAL 6/9 (range 6/4–6/18). Visual acuity was available in 11 (84%) patients, and whereas the median visual acuity in this cohort of mutation carriers is similar to that of all pooled mutation carriers, it is interesting to note that the range is much narrower, indicating better overall acuity. However, it is difficult to draw conclusions, as there were eight mutation carriers in one family (FAM-11), two in another (FAM-1), and a single patient in the final family (FAM-9), with clinical data available for analysis.
Discussion
The BEST1 protein is most strongly expressed in the RPE of the macula compared with other locations in the retina. This has led to speculation that as most cases of VMD are centered around the macula, the disease may be due to a relative insufficiency of wild-type BEST1 and the dominant negative effect of the mutant BEST1.40 The vast majority of BEST1 mutations identified to date are missense, with a significant clustering around amino acid segments 6–30, 80–104, 221–243, and 293–312.16 These regions of the protein are located in or near to the RPE plasma membrane, leading to the hypothesis that this location may be of particular importance for BEST1 function. Interestingly, we identified two novel variants (p.Ile76Val and p.Val81Met) at well-conserved positions, which were predicted by SIFT and Polyphen to be tolerated or benign. This, however, is not inconsistent with previously identified mutations (p.Gly135Ser).41, 42
Despite specific clustering of VMD mutations within BEST1, clear genotype–phenotype correlations are yet to be established for VMD. However, a recent study by a Dutch group purported a severe disease phenotype in one family with a Ala10Val mutation.43 The lack of an overall clear genotype–phenotype correlation is in contrast with BEST1 mutations causing the phenotypes ADVIRC and MRCS, which are generally splice-site mutations causing in-frame deletions or duplications.20 Thus, the reason for the variable penetrance and expressivity of VMD caused by BEST1 mutations, and lack of clear genotype–phenotype correlations, remains controversial. It is interesting to note that in all our pedigrees, with both mutation-positive VMD and clinical VMD, the visual acuity in the better eye reached driving standards (see Table 2; BCVA 6/12 or better) in all but one family (FAM-12, p.Ile76Val). However, this family is thought to also harbour a mutation in the PAX6 gene, as they have phenotypic traits of aniridia. This may then skew the data toward a more severe visual acuity loss.
Genotype–phenotype correlations are likely to be affected by other genetic and environmental factors rather than solely a result of the BEST1 mutation. Various possibilities have been discussed in the literature, including the different physiological effects of the lipofuscin material, incomplete penetrance, environment, and pattern of BEST1 expression in the macula.32, 40, 44
Wabbels and colleagues describe two separate families with reduced penetrance and late disease onset with BEST1 mutations of Ile295del and Asn99Lys.2 Within our cohort, there were no such families with a preponderance of asymptomatic individuals—rather most families had a spectrum of disease severity—with some unaffected mutation carriers balanced by others who were clearly affected (Table 3). As with many other clinical studies of diseases with variable penetrance and expressivity, there was a wide inter- and intra-familial variability in the severity of disease manifestation. More commonly, worse vision was associated with older age.
Several groups have recently investigated the appearance of the OCT in VMD. Spaide et al45 described early lesions with accumulation of yellow material in the outer retina. Later lesions were larger with central clearing of the material and a rim of subretinal deposits at the peripheral border. The material is thought to be unphagocytosed photoreceptor outer segments, with associated subretinal fluid. The accumulation of fluid may be due to the faulty BEST1, resulting in an inefficient RPE pump.45 Atrophic foveas had widespread RPE changes with overlying photoreceptor damage.46 We had similar findings in our cohort with early accumulation of presumed photorecetor deposits with preservation. There have been hypotheses made regarding the stage of the disease, OCT findings, and severity of visual loss. Patients with pseudohypopyon lesions of the macula often have preserved visual acuity, despite significant changes apparent clinically. OCT has shown that in this stage, there may be accumulation of material principally in the subRPE space with preservation of the normal photoreceptor architecture, and therefore relatively normal acuity.45, 46, 47, 48 We compared visual acuity as well as OCT appearance between VMD and clinical VMD patients (Table 3). We found that the clinical VMD group (no BEST1 mutation identified) had thinner CRTs compared with BEST1 mutation carriers (P<0.001). However, this maybe selection bias, as all the patients in this group were the pooled probands without mutations and with no other family members screened.
In the absence of genetic testing, the carrier state may often only be identified in VMD by an abnormal EOG;29 however, some studies have demonstrated preservation of a normal EOG despite the presence of a BEST1 mutation.2, 16, 25, 31 It was not possible to differentiate mutation carriers from non-mutation carriers in our cohort on the basis of EOG alone, and therefore genetic screening remains the mainstay for diagnosis of VMD. We did have a small number of patients (17%) with normal EOGs despite having a BEST1 mutation.
In our cohort of patients, we had 42 mutation carriers and a further 14 patients with a diagnosis of clinical VMD in whom a mutation in BEST 1 was not identified. Interestingly, the phenotypic presentation of the two groups is very similar, and therefore it remains difficult to predict clinically which patients will harbour a BEST1 mutation. Therefore, if VMD is suspected, genetic analysis is required for confirmation.
Limitations of this study include relatively small number of confirmed mutation carriers and failure to detect BEST1 mutations in all pedigrees. One family is thought to have both VMD and aniridia, although this is awaiting genetic confirmation. This would skew the clinical findings as the three patients in this cohort had severe visual loss.
In conclusion, we have demonstrated the genotypic and phenotypic profiles of Australian VMD pedigrees. Median BCVA in the better eye of all patients reached driving standards, which will aid clinicians in counselling the patients regarding prognosis. It remains difficult to differentiate clinically between families with definite VMD and those with presumed VMD, and therefore genetic analysis remains the mainstay for diagnosis. We found no genotype–phenotype correlations in this cohort and further work in this area is required.

References
Best F . Ueber eine hereditaraere Maculaaffektion. Z Augenheilk 1905; 13: 199–212.
Wabbels B, Preising MN, Kretschmann U, Demmler A, Lorenz B . Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol 2006; 244: 1453–1466.
Weber BH, Stohr H, Walker D . A case of nonpenetrance in Best's disease. Am J Ophthalmol 1994; 118: 398–399.
Weber BH, Walker D, Muller B . Molecular evidence for non-penetrance in Best's disease. J Med Genet 1994; 31: 388–392.
Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT . Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev 2008; 88: 639–672.
Marmorstein AD, Cross HE, Peachey NS . Functional roles of bestrophins in ocular epithelia. Prog Retin Eye Res 2009; 28: 206–226.
Marmorstein AD, Kinnick TR . Focus on molecules: bestrophin (best-1). Exp Eye Res 2007; 85: 423–424.
Sun H, Tsunenari T, Yau KW, Nathans J . The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA 2002; 99: 4008–4013.
Graff C, Eriksson A, Forsman K, Sandgren O, Holmgren G, Wadelius C . Refined genetic localization of the Best disease gene in 11q13 and physical mapping of linked markers on radiation hybrids. Hum Genet 1997; 101: 263–270.
Nichols BE, Bascom R, Litt M, McInnes R, Sheffield VC, Stone EM . Refining the locus for Best vitelliform macular dystrophy and mutation analysis of the candidate gene ROM1. Am J Hum Genet 1994; 54: 95–103.
Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998; 19: 241–247.
Stone EM, Kimura AE, Folk JC, Bennett SR, Nichols BE, Streb LM et al. Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum Mol Genet 1992; 1: 685–689.
Kramer F, White K, Pauleikhoff D, Gehrig A, Passmore L, Rivera A et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet 2000; 8: 286–292.
Brecher R, Bird AC . Adult vitelliform macular dystrophy. Eye (London) 1990; 4 (Part 1): 210–215.
Felbor U, Schilling H, Weber BH . Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat 1997; 10: 301–309.
Boon CJ, Klevering BJ, den Hollander AI, Zonneveld MN, Theelen T, Cremers FP et al. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol 2007; 125: 1100–1106.
Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci 2004; 45: 3683–3689.
Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S et al. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Hum Genet 1999; 104: 449–453.
Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet 2009; 85: 581–592.
Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet 2008; 82: 19–31.
Michaelides M, Urquhart J, Holder GE, Restori M, Kayali N, Manson FD et al. Evidence of genetic heterogeneity in MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma) syndrome. Am J Ophthalmol 2006; 141: 418–420.
Reddy MA, Francis PJ, Berry V, Bradshaw K, Patel RJ, Maher ER et al. A clinical and molecular genetic study of a rare dominantly inherited syndrome (MRCS) comprising of microcornea, rod-cone dystrophy, cataract, and posterior staphyloma. Br J Ophthalmol 2003; 87: 197–202.
Clemett R . Vitelliform dystrophy: long-term observations on New Zealand pedigrees. Aust N Z J Ophthalmol 1991; 19: 221–227.
Mohler CW, Fine SL . Long-term evaluation of patients with Best′s vitelliform dystrophy. Ophthalmology 1981; 88: 688–692.
Renner AB, Tillack H, Kraus H, Kramer F, Mohr N, Weber BH et al. Late onset is common in best macular dystrophy associated with VMD2 gene mutations. Ophthalmology 2005; 112: 586–592.
Seddon JM, Sharma S, Chong S, Hutchinson A, Allikmets R, Adelman RA . Phenotype and genotype correlations in two best families. Ophthalmology 2003; 110: 1724–1731.
Cross HE, Bard L . Electro-oculography in Best's macular dystrophy. Am J Ophthalmol 1974; 77: 46–50.
Deutman A . The hereditary dystrophies of the posterior ‘pole. Assen Van Gorcum 1971; 198–299.
Deutman AF . Electro-oculography in families with vitelliform dystrophy of the fovea. Detection of the carrier state. Arch Ophthalmol 1969; 81: 305–316.
Krill AE, Morse PA, Potts AM, Klien BA . Hereditary vitelliruptive macular degeneration. Am J Ophthalmol 1966; 61: 1405–1415.
Testa F, Rossi S, Passerini I, Sodi A, Di Iorio V, Interlandi E et al. A normal electro-oculography in a family affected by best disease with a novel spontaneous mutation of the BEST1 gene. Br J Ophthalmol 2008; 92: 1467–1470.
Caldwell GM, Kakuk LE, Griesinger IB, Simpson SA, Nowak NJ, Small KW et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics 1999; 58: 98–101.
Godel V, Chaine G, Regenbogen L, Coscas G . Best's vitelliform macular dystrophy. Acta Ophthalmol Suppl 1986; 175: 1–31.
Loewenstein A, Godel V, Godel L, Lazar M . Variable phenotypic expressivity of Best′s vitelliform dystrophy. Ophthalmic Paediatr Genet 1993; 14: 131–136.
Ponjavic V, Eksandh L, Andreasson S, Sjostrom K, Bakall B, Ingvast S et al. Clinical expression of Best's vitelliform macular dystrophy in Swedish families with mutations in the bestrophin gene. Ophthalmic Genet 1999; 20: 251–257.
Attebo K, Mitchell P, Smith W . Visual acuity and the causes of visual loss in Australia. The Blue Mountains Eye Study. Ophthalmology 1996; 103: 357–364.
Hee MR, Izatt JA, Swanson EA, Huang D, Schuman JS, Lin CP et al. Optical coherence tomography of the human retina. Arch Ophthalmol 1995; 113: 325–332.
Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W et al. Optical coherence tomography. Science 1991; 254: 1178–1181.
den Dunnen JT, Paalman MH . Standardizing mutation nomenclature: why bother? Hum Mutat 2003; 22: 181–182.
Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM . Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Invest Ophthalmol Vis Sci 2007; 48: 3372–3380.
Bakall B, Marknell T, Ingvast S, Koisti MJ, Sandgren O, Li W et al. The mutation spectrum of the bestrophin protein—functional implications. Hum Genet 1999; 104: 383–389.
Lotery AJ, Munier FL, Fishman GA, Weleber RG, Jacobson SG, Affatigato LM et al. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci 2000; 41: 1291–1296.
Booij JC, Boon CJ, van Schooneveld MJ, ten Brink JB, Bakker A, de Jong PT et al. Course of visual decline in relation to the Best1 genotype in vitelliform macular dystrophy. Ophthalmology 2010; 117: 1415–1422.
Marchant D, Gogat K, Boutboul S, Pequignot M, Sternberg C, Dureau P et al. Identification of novel VMD2 gene mutations in patients with best vitelliform macular dystrophy. Hum Mutat 2001; 17: 235.
Spaide RF, Noble K, Morgan A, Freund KB . Vitelliform macular dystrophy. Ophthalmology 2006; 113: 1392–1400.
Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G, Souied EH . High-definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol 2008; 146: 501–507.
Querques G, Regenbogen M, Soubrane G, Souied EH . High-resolution spectral domain optical coherence tomography findings in multifocal vitelliform macular dystrophy. Surv Ophthalmol 2009; 54: 311–316.
Vedantham V, Ramasamy K . Optical coherence tomography in Best's disease: an observational case report. Am J Ophthalmol 2005; 139: 351–353.
Acknowledgements
We are grateful for the financial support provided by the Clifford Craig Medical Research Trust, the Ophthalmic Research Institute of Australia, and the Peggy and Leslie Cranbourne Foundation. The Centre of Eye Research Australia receives operational and infrastructure support from the Victorian Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Rights and permissions
About this article
Cite this article
Cohn, A., Turnbull, C., Ruddle, J. et al. Best's macular dystrophy in Australia: phenotypic profile and identification of novel BEST1 mutations. Eye 25, 208–217 (2011). https://doi.org/10.1038/eye.2010.180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2010.180
Keywords
This article is cited by
-
Spectrum of variants associated with inherited retinal dystrophies in Northeast Mexico
BMC Ophthalmology (2024)
