
Abstract
Aim To describe the occurrence of bilateral primary choroidal melanoma in four patients.
Methods All patients attending the Liverpool Ocular Oncology Centre with uveal melanoma between January 1993 and February 2002 were identified, and those with bilateral primary choroidal melanoma were reviewed. Their presentation and management are described.
Results Four patients, all female, were identified. Patient 1 presented with a right juxtapapillary melanoma at the age of 64, which was treated with krypton laser and endoresection, and then when aged 73 required proton beam radiotherapy for a melanoma in her left eye. Patient 2 presented at the age of 82 with bilateral choroidal melanomas and underwent simultaneous bilateral plaque radiotherapy. Patient 3 presented with bilateral choroidal melanomas at the age of 75 and was treated initially with bilateral proton beam radiotherapy. Patient 4 was treated at the age of 54 with right plaque radiotherapy for a choroidal melanoma, and 3 years later needed plaque radiotherapy for a melanoma in her other eye.
Conclusion Bilateral choroidal melanoma is possible and should be a consideration in the continuing management of patients with choroidal melanoma.
Similar content being viewed by others
Introduction
Uveal melanoma is usually a sporadic disease, although it may rarely be associated with ocular melanocytosis, neurofibromatosis type I and familial atypical mole and melanoma syndrome.1 Bilateral uveal melanoma is exceedingly rare and only isolated cases are usually reported. Since 1930, there have been less than 50 well-documented cases described.2
The aims of this study were to describe the presentation and outcome of four patients with bilateral uveal melanoma.
Materials and methods
A search of the Liverpool Ocular Oncology Centre (LOOC) database revealed four patients with bilateral uveal melanoma, who had been treated between January 1993 and February 2002. The database contained information on all patients presenting to LOOC during this period. The clinical data and diagnosis were simultaneously recorded in proformas in the casenotes and computerized by the ophthalmologist at the time the patients were seen, using a computer terminal in the clinic. Entries were subsequently checked by a data manager, who ensured that the records were complete and correct.
At each consultation, a systematic history was taken according to a proforma. The corrected visual acuity was recorded, using a Snellen chart. Both eyes were examined by slit-lamp biomicroscopy and indirect ophthalmoscopy. B-scan ultrasonography was performed by the ophthalmologist, using a high-frequency probe for anterior segment tumours. Colour photographs were also taken at each visit.
In all patients, the diagnosis was established by one clinician (BED) and was based on the clinical and ultrasonographic findings. This was confirmed by histology in four tumours undergoing biopsy, local resection, or enucleation.
Results
Four patients with bilateral uveal melanoma were identified. The patients, all females, were between 54 and 75 years of age at the time of their initial presentation. The diagnosis was made simultaneously in two cases, and in the other two the second melanoma was diagnosed after intervals of 9 and 3 years, respectively. The case reports are given below.
Case 1
In January 1993, a healthy 66-year-old woman was referred to LOOC for follow-up. In July 1991, she had been treated by the same ophthalmologist (BD) in Glasgow for a juxtapapillary choroidal melanoma in her right eye with low-dose, long-duration krypton laser photocoagulation3 followed by endoresection.4 On examination in February 1994, a small choroidal lesion was noted in the left eye, approximately 2DD inferotemporal to the fovea, and was thought to be a naevus. The tumour had not been noted previously, in numerous follow-up examinations in Glasgow, Liverpool, and at her own hospital. The patient was also being treated for ocular hypertension by her primary ophthalmologist, who commenced Teoptic 2% b.i.d. OU in March 1994 because of an intraocular pressure of 26 mmHg in each eye.
In April 1998, an amelanotic choroidal tumour had grown in the left eye, corresponding to the previously noted pigmented tumour. Visual acuity was 6/6. On ultrasonography, the tumour had a largest basal diameter (LBD) of 5.4 mm and a thickness of 1.2 mm. The patient was initially kept under observation, without treatment, but in August 2000 the tumour was noted to have grown, developing a thickness of 2.2 mm and a basal diameter of 11.4 mm (Figure 1). The patient was therefore treated with proton beam radiotherapy. When last reviewed, in June 2002, the patient was healthy, with no sign of systemic metastatic disease. The right eye had a visual acuity of 1/60, moderate cataract, and a surgical choroidal coloboma involving optic disc and macula, with no evidence of local tumour recurrence. The visual acuity in the left eye was 6/12 and the tumour thickness was approximately the same at 2.4 mm. There was some exudation from the tumour and therefore transpupillary thermotherapy was performed to prevent macula oedema.

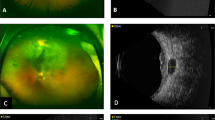
Case 1. Fundus photograph of the left eye. A choroidal tumour is visible inferotemporal to the macula (September 2000).
Case 2
A healthy 82-year-old female was referred to the LOOC in September 2000 with a choroidal tumour in her right eye. On examination, the right eye had a visual acuity of 6/12, moderate cataract, and a pigmented choroidal tumour superonasally near the equator. On ultrasonography, the tumour had an LBD of 7.5 mm and was 2.2 mm thick. The left eye had a visual acuity of 6/12, conjunctival melanosis, and a moderate cataract. On ophthalmoscopy, a diffuse pigmented choroidal tumour was discovered in the superonasal quadrant, also in the equatorial region. On ultrasonography, it had an LBD of 14.5 mm and a thickness of 1.4 mm.
The patient was treated with bilateral simultaneous ruthenium plaque radiotherapy. When last reviewed in May 2001, the patient was healthy with no sign of systemic metastatic disease. Visual acuity was 6/18 in the right eye and 6/12 in the left, and both tumours had diminished in thickness to 1.0 mm.
Case 3
A healthy 75-year-old woman was referred in September 2000 to the LOOC with bilateral choroidal tumours. She had had a malignant melanoma removed from her left forearm in 1996. On examination, the right eye had a visual acuity of 6/24, some pigmentation beneath the superonasal bulbar conjunctiva, moderate cataract, and a pigment ciliary body tumour in the superonasal quadrant. On ultrasonography, the tumour had an LBD of 13.0 mm and was 9.5 mm thick. The left eye had a visual acuity of 6/9, mild cataract, and a pigmented choroidal tumour immediately inferior to the left fovea. On ultrasonography, the tumour measured 13.0 mm in LBD and was 5.1 mm thick. The patient was treated with bilateral proton beam radiotherapy, with a shave biopsy of the right eye and a choroidal biopsy of the left eye tumour confirming the diagnosis of bilateral choroidal melanoma, with a cell type of ‘Spindle B’.
When reviewed in January 2001, the patient was healthy and free of systemic disease. The visual acuity in the right eye was 6/36 and the tumour had decreased in thickness to 8.73 mm. The visual acuity in the left eye was no perception of light and the eye was painful, because of neovascular glaucoma. On ultrasonography, the tumour was 4.0 mm thick. Enucleation was performed, providing symptomatic relief and confirming the diagnosis of choroidal melanoma (Figure 2).

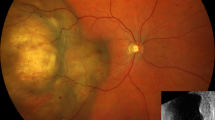
Case 3. Enucleation specimen of the left eye. There is recurrent choroidal melanoma present near the bottom of the specimen (January 2001).
Case 4
A healthy 54-year-old female was referred to the LOOC in November 1994. She had been under the care of the same ophthalmologist (BD) for several years at the Tennent Eye Institute, Glasgow, with the diagnosis of suspicious pigmented choroidal tumour in the right eye. On examination the visual acuity was 6/5 in each eye. Both anterior segments were normal and the left fundus was healthy. There was a pigmented choroidal tumour in the superotemporal quadrant of the right eye, with some overlying lipofuscin, which had not been present when previously examined in Glasgow. On ultrasonography, the lesion had an LBD of 5.5 mm and was 1.8 mm thick, unchanged when compared with previous measurements. The patient was treated with a right 15 mm ruthenium plaque because of the recent appearance of lipofuscin.
On examination in January 1997, a new pigmented pars plana tumour was detected in the inferotemporal quadrant of the left eye. On ultrasonography, this measured 8 mm in LBD and was 2 mm thick. Visual acuity remained 6/6 in each eye. This was now treated with a ruthenium plaque after confirming the diagnosis by choroidal biopsy. This was complicated by wound dehiscence, which was successfully treated with an autologous scleral graft. A small prolapsed portion of tumour was excised and the diagnosis reconfirmed.
When last reviewed in January 2002, the visual acuity in both eyes was 6/6 and tumours had decreased in thickness to 0.4 and 0 mm in the right and left eyes, respectively. The patient remains healthy and free of systemic metastatic disease.
Discussion
Between January 1993 and March 2002, 1835 patients with uveal melanoma were seen at LOOC. Four of these patients, all from the British Isles, were diagnosed with bilateral uveal melanoma over this time period. In two cases, both tumours were present at the time of the initial diagnosis and in the other two the tumour developed in the fellow eye at a later date. With particular regard to the two patients who had simultaneous involvement with uveal melanoma, the possibility that this did not represent bilateral primary disease must be considered. Metastasis of uveal melanoma from one eye to the other has been described,5 as having ocular metastases from cutaneous malignant melanoma.6 However, we believe these possibilities to be unlikely as at the most recent follow-up there is no evidence of metastatic disease in any patient, and only one of our cases had a history of cutaneous malignant melanoma (case 3). Neither did any case have another predisposing condition such as oculodermal melanocytosis.
Singh et al7 estimated that the probability of any one individual developing uveal melanoma during his lifetime is 7.36 × 10−5. Since the patients nearly all have only one eye affected, and since left and right eyes are equally likely to be affected, the lifetime probability of left-eye melanoma=the probability of right-eye melanoma=3.68 × 10−5, which is half of Singh's figure. If the occurrences in left and right eyes were mutually independent (statistically unrelated), we would expect the lifetime probability of bilateral uveal melanoma to be the square of this figure, that is, 1.35 × 10−9; and within the white population of 5 × 107 in the United Kingdom, the number of patients expected to develop bilateral melanoma in their lifetime would therefore be only 0.068. If this is subject to a Poisson-distributed sampling error, the probability of observing four such people would be less than 0.00001.
The discrepancy becomes even greater when we consider that the figure of 1.35 × 10−9 is an upper limit for the prevalence of bilateral uveal melanoma at any one time, as each individual will be affected by uveal melanoma for only part of his life. As we have four such cases, all still alive, the prevalence of bilateral uveal melanoma is much too great to be compatible with the assumption that the pathology arises independently in left and right eyes by random chance.
The possibility of bilateral disease should be taken into account when considering enucleation vs conservative treatment. Similarly, in patients with a history of choroidal melanoma, it is important to examine the other eye and carefully observe any choroidal naevus in case of malignant transformation.
References
Singh AD, Donoso LA . Genetic aspects of uveal melanoma. Int Ophthalmic Clin 1993; 33(3): 47–52.
Singh AD, Shields CL, Shields JA, De Potter P . Bilateral primary uveal melanoma. Ophthalmology 1996; 103: 256–262.
Bornfeld N . Laser treatment of choroidal melanoma. In: Ryan SJ (editor-in-chief). Retina. Mosby: St Louis, 2001, pp 785–794.
Damato B, Groenewald C, McGalliard J, Wong D . Endoresection of choroidal melanoma. Br J Ophthalmol 1998; 82: 213–218.
Singh AD, Shields JA, Shields CL, Sato T . Choroidal melanoma metastatic to the contralateral choroid. Am J Ophthalmol 2001; 132: 941–943.
Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE . Survey of 520 eyes with uveal metastasis. Ophthalmology 1997; 104: 1265–1276.
Singh AD, Wang MX, Donoso LA, Shields CL, De Potter P, Shields JA et al. Familial uveal melanoma—III: is the occurrence of familial uveal melanoma coincidental? Arch Ophthalmol 1996; 114: 1101–1104.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hadden, P., Damato, B. & McKay, I. Bilateral uveal melanoma: a series of four cases. Eye 17, 613–616 (2003). https://doi.org/10.1038/sj.eye.6700435
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700435
Keywords
This article is cited by
-
Genetik des Aderhautmelanoms
Spektrum der Augenheilkunde (2009)
