
Abstract
Paget’s disease of bone (PDB) is a common metabolic bone disease that is characterized by aberrant focal bone remodeling, which is caused by excessive osteoclastic bone resorption followed by disorganized osteoblastic bone formation. Genetic factors are a critical determinant of PDB pathogenesis, and several susceptibility genes and loci have been reported, including SQSTM1, TNFSF11A, TNFRSF11B, VCP, OPTN, CSF1 and DCSTAMP. Herein, we report a case of Chinese familial PDB without mutations in known genes and identify a novel c.163G>C (p.Val55Leu) mutation in FKBP5 (encodes FK506-binding protein 51, FKBP51) associated with PDB using whole-exome sequencing. Mutant FKBP51 enhanced the Akt phosphorylation and kinase activity in cells. A study of osteoclast function using FKBP51V55L KI transgenic mice proved that osteoclast precursors from FKBP51V55L mice were hyperresponsive to RANKL, and osteoclasts derived from FKBP51V55L mice displayed more intensive bone resorbing activity than did FKBP51WT controls. The osteoclast-specific molecules tartrate-resistant acid phosphatase, osteoclast-associated receptor and transcription factor NFATC1 were increased in bone marrow-derived monocyte/macrophage cells (BMMs) from FKBP51V55L mice during osteoclast differentiation. However, c-fos expression showed no significant difference in the wild-type and mutant groups. Akt phosphorylation in FKBP51V55L BMMs was elevated in response to RANKL. In contrast, IκB degradation, ERK phosphorylation and LC3II expression showed no difference in wild-type and mutant BMMs. Micro-CT analysis revealed an intensive trabecular bone resorption pattern in FKBP51V55L mice, and suspicious osteolytic bone lesions were noted in three-dimensional reconstruction of distal femurs from mutant mice. These results demonstrate that the mutant FKBP51V55L promotes osteoclastogenesis and function, which could subsequently participate in PDB development.
Similar content being viewed by others

Introduction
Paget’s disease of bone (PDB (MIM 167250)) is a common skeletal disorder that is characterized by abnormal focal bone remodeling. It usually affects one or more bones in patients with multiple constitutive symptoms including bone pain, deformity, pathological fractures, deafness and secondary osteoarthritis.1 In European countries, PDB is the second most frequently diagnosed chronic bone disorder after osteoporosis, and it affects ~5% of women and 8% of men in the UK by the age of 80.2 However, PDB is rarely diagnosed in Asian populations; for example, the prevalence of PDB in Japan is only ~0.00028%.3
Although the exact pathogenesis of PDB is still unclear, genetic factors are known to be important in PDB development. Mutations in the SQSTM1 gene, which encodes p62, have been shown to be widely involved in PDB development and in the severity of the disease phenotype. Among these mutations, p62P392L is the most common; it has been shown to be involved in ~20–50% of cases of familial PDB and in 16% of cases of sporadic PDB.4 In addition to SQSTM1, candidate gene approach and genome-wide association studies have identified several other PDB susceptibility genes and loci; these include TNFRSF11A, OPTN, CSF1, TNFRSF11B, VCP and DCSTAMP.5, 6, 7, 8 Environmental factors such as virus infection, low-calcium diet, and exposure to pets and toxins have also been proposed to contribute to the etiology of PDB. Although still controversial, some consider paramyxovirus infection, especially measles virus infection, to be a potential risk factor in PDB development.9, 10, 11 Recently, a marked decline in the prevalence of PDB in some regions has been noted, a finding that suggests a role for environmental factors in PDB development.9
Osteoclast dysfunction is the primary defect in PDB.12 Osteoclasts in pagetic bone lesions display increased size, contain multiple nuclei and possess characteristic intranuclear inclusion bodies.13 Osteoclasts derived from the peripheral blood monocytes of patients with PDB are more sensitive than osteoclasts from non-PDB patients to factors that induce bone resorption, including the 1,25-dihydroxyvitamin D3 and receptor activator for nuclear factor κB (NFκB) ligand (RANKL).14 RANKL is a key cytokine that induces osteoclastogenesis.15 The binding of RANKL to its receptor activates signaling pathways including the NF-κB, Akt and MAPK pathways; these pathways ultimately activate NFATC1 and initiate osteoclastogenesis.15 Thus, aberrant regulation of specific signaling pathways is an important component of PDB etiology that may contribute to osteoclast dysfunction and disease development.
In this study, we recruited an extremely rare case of Chinese familial PDB, and genetic disorders in this familial case were studied. No mutations in previously reported genes were found in this family. Instead, a novel missense mutation in FKBP5 was identified using whole-exome sequencing. FKBP5 encodes the FKBP51 protein, which is a regulator of NF-κB activation and Akt phosphorylation.16, 17, 18 Given the correlation between the function of FKBP51 and osteoclastogenesis-related signaling pathways, we hypothesized that the FKBP5 mutation might cause PDB by influencing osteoclast formation and biological function.
Materials and methods
Studied family
Three patients in the studied family were evaluated at the Third Hospital of Jinan, and all patients were clinically diagnosed based on their clinical features and characterized digital radiography (DR) examination findings in bone.1 This study was approved by the ethics committee of Shandong Provincial Hospital Affiliated to Shandong University, and written informed consent was obtained from all participants.
DNA sequencing
Genomic DNA was extracted from peripheral blood samples of all family members using a blood DNA extraction kit (Qiagen, Duesseldorf, Germany). To determine whether alterations were present in known PDB susceptibility genes in this family, we sequenced the SQSTM1, TNFRSF11A, TNFRSF11B, VCP, OPTN, CSF1 and DCSTAMP genes of all family members using the polymerase chain reaction (PCR) followed by Sanger dideoxy sequencing on an ABI 3100 Genetic Analyzer.
Whole-exome sequencing and candidate gene screening
We performed whole-exome sequencing of three patients (II3, II6 and II11) and their two healthy siblings (II4, II8). Exome enrichment was achieved using the Agilent Sure-Select Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed on an Illumina instrument using paired-end 101-bp reads at a sequencing depth of × 100. The sequence reads were mapped to the Human Reference Genome; then, single-nucleotide variants (SNVs) annotation was performed using the Burrows–Wheeler Aligner and Samtools. SNVs with one of the following conditions, a quality score higher than 30, a quality score-to-depth ratio >2.5, or a map quality score >25, were chosen for further analysis.
Candidate mutations that co-segregated with PDB were filtered from the set of reversed SNVs according to the following filtering conditions: absence from the Single-Nucleotide Polymorphism Database (dbSNP), non-synonymous mutations located in the gene-coding area, existence in all affected patients, and absence from both unaffected sibling controls.
FKBP5 gene mutation verification and function prediction
Five SNVs that co-segregated with PDB were identified using the whole-exome sequencing assay. Of these, the c.163G>C mutation (p.Val55Leu) in exon 4 of FKBP5 was selected as the candidate because FKBP5 encodes FKBP51, which is functionally associated with osteoclast physiology. To verify this mutation in the patients and determine the status of this mutation in the Han Chinese population (that is, to determine whether it is an undefined single-nucleotide polymorphism (SNP)), we sequenced PCR-amplified segments of FKBP5 exon 4 in family members and in 200 Han Chinese controls using standard Sanger sequencing methodology. Function prediction of the mutant FKBP51 was performed using PloyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and PyMol (DeLano Scientific LLC, San Carlos, CA, USA).
Functional assay of mutant FKBP51 in cell experiments
The function of the mutant FKBP51V55L in the regulation of NF-κB transcriptional activity was measured by luciferase experiments in HEK293 cells. Briefly, pcDNA3.1-FKBP51, pcDNA3.1-FKBP51V55L or control plasmid were co-transfected with the PGL4.32[luc2p/NF-κB-RE/Hygro] luciferase reporter vector and the PRL-TK plasmid into HEK293 cells. Twenty-four hours after transfection, the cells were treated with 20 ng ml−1 tumor necrosis factor-alpha for 4 h and processed for luciferase assays using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
To detect Akt phosphorylation, wild-type or mutant FKBP51-overexpressing HEK293 cells were serum-starved for 12 h and then incubated with lipopolysaccharide (1 μg ml−1, Sigma, St Louis, MO, USA) for 15 min. Total proteins were immediately extracted from the cells, and Akt phosphorylation was detected by western blotting. A rabbit antibody to phospho-Akt (Ser473) and a rabbit antibody to Akt were obtained from Cell Signaling Technology (Danvers, MA, USA). A rabbit antibody to GAPDH and a goat antibody to rabbit IgG were obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Monoclonal U937 cell lines expressing wild-type or mutant FKBP51 were serum-starved for 12 h and then activated by RANKL (100 ng ml−1, R&D Systems, Minneapolis, MN, USA) for 15 min. The kinase activity of Akt in these cells was then measured using the KinaseSTAR Akt Activity Assay Kit (BioVision, Milpitas, CA, USA). The kinase activity of Akt was finally determined by examining the amount of exogenous phosphorylated GSK-3α using western blotting.
Generation of FKBP51V55L KI transgenic mice
All animal studies were approved by the Institutional Animal Care and Use Committee of Shandong Provincial Hospital affiliated to Shandong University. Animal handling and experiments were performed according to the local legislation titled ‘Regulations for The Administration of Affairs Concerning Experimental Animals.’
To generate KI mice carrying the FKBP51V55L mutation in the endogenous mouse FKBP5 gene (GenBank accession number: NM_010220.4), a target vector containing homology arms, a Neo cassette flanked by LoxP sites, a c.163G>C mutation in exon 3 that results in generation of the V55L mutation in the 3′ homology arm, and a DTA cassette in the 3′ flank was created. The homology arms were generated by PCR using the BAC clones RP23-77G4 and RP23-238B10 from the C57BL/6J library as a template. The c.163G>C mutation was created by site-directed mutagenesis using the TaKaRa MutanBEST Kit (TaKaRa Biotechnology, Dalian, China), and this method was used to insert two AflII and two HindIII restriction enzyme sites, both of which spanned the target sequence, to facilitate the identification of the targeting event. The targeting construct was electroporated into C57BL/6 ES cells, and G418-resistant clones were picked. These clones were further characterized using PCR assays with the primers 3′ PCR_F and 3′ PCR_R, by which positive clones would produce a 3.6-kb fragment. In addition, positive ES clones were identified for correct integration using Southern blotting of AflII- and HindIII-digested genomic DNA, and these positive clones were sequenced for the c.163G>C mutation in exon 3 using a pair of sequencing primers. Cells from the positive clones were injected into recipient blastocysts obtained from pregnant C57BL/6 female mice. The chimeric offspring of the mice were mated with CMV-Cre-expressing C57BL/6 mice, and the offspring of these mice were tested for germline transmission of the targeting sequence and for excision of the neomycin cassette by PCR using a pair of primers that flank the inserted LoxP site (F1 and R1; Figure 3a). Positive pups in which a successful deletion of the neomycin cassette was identified were further genotyped by Sanger sequencing using a pair of primers that flank the mutation site: sense: 5′-CCTCGAGGCATGCGATAATCTAGCT-3′; antisense: 5′-GGCAAATGGCTTCTTTCTGTCATG-3′. The mouse colony was maintained by breeding heterozygotes; the wild-type and homozygous mice used in this study are littermates.
Osteoclast formation and TRAP staining
Osteoclast precursors were prepared as previously described.19 In brief, mouse bone marrow cells were isolated from the femurs and tibiae of 7-week-old mice. The cells were cultured in α-minimum essential medium (Life Technologies, Grand Island, NY, USA) containing 10% fetal bovine serum with macrophage colony-stimulating factor (M-CSF) (5 ng ml−1) for 12 h in 100-mm-diameter dishes (Corning, Corning, NY, USA; 1 × 107 cells per 10 ml per dish). After 12 h, nonadherent cells were collected and cultured in 48-well plates (Corning; 1.5 × 105 cells per well) with M-CSF (30 ng ml−1). After 3 days, the floating cells were removed, and the attached cells were used as bone marrow-derived monocyte/macrophage cells (BMMs). The BMMs were cultured in the presence of varying concentrations of RANKL and M-CSF (50 ng ml−1) for 3 days. The cells were then fixed and stained for tartrate-resistant acid phosphatase (TRAP), as described in the manual provided by the manufacturer (387A, Sigma-Aldrich, Steinheim, Germany). TRAP-positive giant cells containing more than two nuclei were identified as osteoclasts and counted.
Pit formation assay
Bone marrow cells (2 × 105 cells per well) were plated on bovine bone slices (4 mm × 6 mm) that had been placed in 48-well plates. The bone marrow cells were first cultured with M-CSF (30 ng ml−1) for 3 days. The slices were then washed and further cultured with RANKL (100 ng ml−1) and M-CSF (50 ng ml−1) for an additional 7 days, with the medium changed every 3 days. The slices were then cleaned by ultrasonication in 0.25 M NH4OH buffer and stained with 1% toluidine blue. Resorption pits were photographed under microscopic examination, and the areas of the pits were calculated using Image-Pro Plus (Version X; Media Cybernetics, Silver Springs, MD, USA).
Analysis of osteoclast-specific gene expression
BMMs were cultured in six-well plates with RANKL (100 ng ml−1) and M-CSF (50 ng ml−1) for the indicated times. The cells were then lysed with TRIzol (Life Technologies), and RNA was extracted. RNA (1 μg) was reverse-transcribed with the Transcriptor First-Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) using random hexamer primers in a final volume of 20 μl. Relative quantification of real-time PCR reactions was performed, and the resulting data were analyzed using Roche LightCycler 480. Subsequently, the analysis results were visualized by agarose gel electrophoresis. Each reaction contained 10 μl of SYBR Green I Master (Roche Applied Science), 1 μl of cDNA, 1 μl of primer pairs (10 μM) and 8 μl of distilled H2O. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used as a reference control.
Transcription factors and signaling pathways detection
BMMs were cultured in six-well plates and induced with RANKL (100 ng ml−1) and M-CSF (50 ng ml−1) for the indicated amounts of time. Then, c-fos and NFATC1 expression was detected using western blotting. To measure signaling pathway activation, BMMs were serum-starved for 4 h before exposure to RANKL for the indicated times; then, the phosphorylation of Akt and Erk, and degradation of IκB were detected by western blotting. For LC3 expression, BMMs were cultured in six-well plates in the presence of RANKL (100 ng ml−1) and M-CSF (50 ng ml−1) for the indicated number of days, and the LC3 expression levels were determined using western blotting. Rabbit antibodies to c-fos, NFATC1 and IκB were obtained from Abcam (Cambridge, MA, USA); rabbit antibodies to pErk, Erk and LC3 were obtained from Cell Signaling Technology.
Micro-CT analysis
The hind limbs of 10-month-old FKBP51WT and FKBP51V55L male littermates (three wild-type and three homozygous littermates) were obtained and fixed in 4% formalin-buffered saline. Micro-CT analysis was performed using a Siemens Inveon micro-CT system (Siemens Medical Solutions, Knoxville, TN, USA). For the assessment of bone morphometry, bilateral femurs and tibiae of mice were dissected free of most soft tissues, and the femur and proximal tibia were scanned at an effective pixel size of 9.08 μm (60 kV, 220 μA). Two-dimensional images and three-dimensional (3D) reconstruction of femurs were performed using the Inveon Acquisition Workplace. Trabecular bone parameters were analyzed using the Inveon Research Workplace. Briefly, we measured a stack of slices within a 0.5-mm step length that began at the slice 0.5 mm distal to the growth plate and ended at the slice 1 mm distal to the growth plate. The cortical thickness 1 mm distal to the growth plate was also measured.
Statistical analysis
The experimental results were statistically analyzed using Student’s t-test. P<0.05 was considered statistically significant.
Results
The studied family
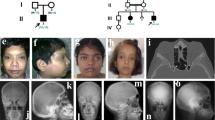
The pedigree of the studied family is shown in Figure 1e. Patient II3, the proband of the family, was diagnosed with PDB at 61 years of age when she came to the hospital seeking treatment for lung discomfort. Marked pagetic bone lesions in her humeri were noticed during lung X-ray examination, and subsequent whole body skeletal scanning by DR showed that her skull, humeri, vertebrae, pelvis and femurs were affected by PDB. Typical pagetic bone lesions in the affected bones of this patient are shown in Figures 1a–d. Serum alkaline phosphatase (ALP) (2637 IU l−1, normal range: 23–140 IU l−1) at the time of diagnosis was remarkably elevated in this patient. Cervical and limb weakness and body height reduction were also noted. No other clinical symptoms or secondary complications such as fracture, osteoarthritis, compression neuropathy or high-output heart failure were noted.

Typical images of bone lesions in the proband and within the pedigree of the studied family. (a) Digital radiography (DR) of the proband’s skull shows thickening of the outer and inner plates of the cranial bones and a ‘cotton wool’ appearance caused by irregular areas of sclerosis. (b) CT bone scanning of the skull shows confirmed bone expansion, cortical bone thickening, widening of the diploё, and irregular areas of sclerosis (the white arrows indicate irregular areas of sclerosis). (c) Radiography of the proband’s pelvis shows mixed areas of lucency and sclerosis and a trabecular pattern of coarsening and accentuation, as well as thickening of the femoral cortices. (d) DR image of the affected humerus shows marked cortical bone thickening and expansion. (e) Pedigree of the affected family. Squares denote male members, and circles represent female members. The symbols crossed by oblique lines represent deceased family members. The black solid symbols indicate affected family members, and gray solid symbols represent offspring who harbor the FKBP5 missense mutation. The dark arrow indicates the proband of this pedigree.
Once the proband was diagnosed, her family members were invited to participate in the study and were screened for PDB. Two siblings (II6 and II11) of this proband were diagnosed with PDB. Patient II6 was 54 years of age when she was diagnosed. DR examination showed that her skull, humeri, vertebrae, pelvis and femurs were affected and that her serum ALP (3337 IU l−1) at the time of diagnosis was elevated. This patient’s main clinical symptoms were low back pain, cervical and limb weakness, and slight spinal deformity. The patient claimed that cervical and limb weakness had appeared ~2 years prior to diagnosis. No other complications were found. Patient II11 was 45 years of age when he was diagnosed. DR examination showed that his skull, vertebrae and pelvis were affected and that his serum ALP (1851 IU l−1) at the time of diagnosis was elevated. However, sclerosis and bone expansion in this patient were mild compared with those in the other two patients. At the beginning of the study, this patient reported no significant symptoms.
The patients and some of the patients’ family members were again invited for further examination and treatment ~2 years ago. DR examination confirmed that there were no new affected bones in the patients. However, the spinal deformity of patient II6 had become more severe. A body height reduction of ~9 cm was noted in patient II11, who complained of cervical weakness. Magnetic resonance examination showed that the first, second and third lumbar vertebrae of this patient were severely compressed and presented typical double-sided depressed deformities (not shown). The serum ALP levels of these patients demonstrated progressive ALP elevation (Table 1). Intravenous infusion of zoledronic acid (Aclasta, Novartis Pharma Schweiz AG, Switzerland) at 5 mg per year was used for each patient. After 2 years of treatment, the symptoms of patients II3 and II11, including cervical and limb weakness, and body height reduction, were significantly remitted; the serum ALP levels of these two patients also significantly decreased (Table 1), and the calcium and phosphorus homeostasis of the patients was normal. Patient II6 did not return for examination. Five offspring of the patients were also examined, and none of them showed pagetic changes. Detailed information on the patients and their offspring, and the results of the latest examination are provided in Table 1.
No mutations in previously reported disease-related genes were found in the family
Except for the presence of many SNPs, no mutations in SQSTM1, TNFRSF11A, TNFRSF11B, VCP, OPTN, CSF1 and DCSTAMP genes were found in this family. All identified SNPs in these genes are listed in Table 2.
Whole-exome sequencing targeted a missense point mutation in FKBP5 gene as candidate
A total of 245 982 SNVs were detected in five samples using WES. Considering the extremely low prevalence of PDB in China, we hypothesized that disease-causing genetic variants would not likely include known SNPs in the general population. Hence, only missense mutations that did not exist in dbSNP and that co-segregated with disease (existed in all patients and were absent from their normal siblings) were considered as candidates. Using this approach, five missense mutations in five genes (FKBP5, ACBD4, MYO7B, AKNA and CCL4L1) were identified (Table 3). The mutations in ACBD4 and MYO7B were previously reported in the Exome Aggregation Consortium (ExAC) database; the mutations in FKBP5, AKNA and CCL4L1 show no records in the ExAC database.
The mutation in FKBP5 was selected for further investigation because of the important biological functions of FKBP5. FKBP51 has been shown to regulate the NF-κB and Akt pathways, both of which are involved in osteoclastogenesis.15, 16, 18 These confirmed functions of FKBP51, which may influence osteoclast generation, made the FKBP5 mutation a good candidate for a mutation associated with the development of PDB. The other four genes were initially excluded because their previously described functions are unlikely to be associated with this disease.
FKBP5 gene mutation is verified and is predicted to be deleterious to Akt signaling
The identified FKBP5 mutation (c.163G>C) is located within exon 4 of the FKBP5 gene. None of the 200 Han Chinese individuals examined had this mutation, which confirmed that it is a novel single-nucleotide mutation rather than a polymorphism. Genotyping of the c.163G>C mutation confirmed that all of the patients in the study were GC heterozygotes. Five of the offspring of the patients (III3, III13, III15, III17 and IV2) harbored the same mutation as the patients, and they were GC heterozygotes as well (Figure 2a). Figure 1e shows the offspring of patients who inherited this heterozygous mutation.

Identification and determination of the effects of the FKBP5 gene mutation. (a) Sanger sequencing data of codons 160–168 in exon 4 of FKBP5 in a healthy member and a patient, illustrating the presence of an obvious c.163G>C heterozygous mutation in the patient (indicated by the orange arrow). (b) Amino-acid sequence alignment comparing the sequence of FKBP51 in 13 vertebrates. The alignment illustrates the highly conserved nature of the V55 residue in FKBP51. Positions identical to Homo sapiens are highlighted in blue. (c) Three-dimensional structure of the FK1 domain of the wild-type and mutant FKBP51 proteins. The yellow arrow indicates the position of the V55L mutation in which valine is replaced by leucine. (d) Relative luciferase activity of transfected HEK293 cells. There is no significant difference in the luciferase activity of cells transfected with wild-type and mutant FKBP51 (P=0.3624). The data are representative of at least three independent experiments. (e) Western blot showing that both wild-type and mutant FKBP51 are overexpressed in transfected HEK293 cells. (f) A representative western blotting result of Akt phosphorylation in transfected HEK293 cells; the blot shows a higher pAktser473 level in the cells transfected with mutant FKBP51 than in cells transfected with the wild-type protein. (g) Band intensity ratios of phosphorylated AktSer473 to total Akt determined by western blotting. The result shows that the mutant FKBP51 increased Akt phosphorylation in HEK293 cells (P=0.0075). The data are representative of at least three independent experiments. **P<0.01. (h) Phosphorylated GSK-3α detected by western blotting. The blot shows the phosphorylation levels of exogenous GSK-3α phosphorylated by endogenous Akt, which reflects the kinase activity of Akt, in U937 cells expressing wild-type and mutant FKBP51. This result proves that Akt kinase activity in mutant FKBP51-transfected U937 cells is higher than in cells expressing the wild-type protein.
FKBP51 has three functional domains: two consecutive FKBP12-like domains (the FK1 and FK2 domains) and a three-unit repeat of the tetratricopeptide repeat domain.20 The c.163G>C mutation results in a p.Val55Leu replacement at a highly conserved position within the FK1 domain (Figure 2b). The 3D structure of mutant FKBP51V55L showed that the V55L mutation lies within the first N-terminal beta strand of FKBP51, precisely on the surface of the FK1 functional pocket (Figure 2c).
Assays showed that the luciferase activity of HEK293 cells transfected with wild-type or mutant FKBP51 did not significantly differ (Figure 2d). However, the Akt phosphorylation level at Ser473 was increased in cells transfected with mutant FKBP51 compared to that in cells transfected with the wild-type control (Figures 2e–g). The phosphorylation level of exogenous GSK-3α, which was phosphorylated by the total Akt extracted from mutant FKBP51-expressing U937 cells, was increased compared with the wild-type group, thus demonstrating that Akt kinase activity is increased in U937 cells expressing the mutant FKBP51 protein (Figure 2h).
Osteoclast formation and bone resorption activity
FKBP51V55L KI transgenic mice were created and used to study osteoclast differentiation and bone resorption activity induced by RANKL in vitro. When the concentration of RANKL was low (30 ng ml−1), osteoclasts derived from wild-type and FKBP51V55L mutant BMMs showed no significant differences. When the concentration of RANKL was increased to 70 or 150 ng ml−1, the number of osteoclasts derived from both FKBP51WT and FKBP51V55L BMMs was increased compared with the low concentration induction groups, and the number of osteoclasts derived from FKBP51V55L BMMs were significantly increased compared with that from FKBP51WT BMMs at the same RANKL induction concentration (Figures 3b and d). The bone resorption function of osteoclasts derived from FKBP51V55L transgenic mice and control mice was also detected by the pit forming assay. We found that the mean area of resorption pits formed by osteoclasts derived from FKBP51V55L transgenic mice was larger than that of resorption pits formed by osteoclasts from control mice (Figures 3c and e).

The FKBP51V55L mutation promotes osteoclastogenesis and enhances osteoclast bone resorption activity. (a) Genotyping strategy for the generation of FKBP51V55L KI mice. The upper panel shows the structure of the murine FKBP5 gene; the location of codon 55 in exon 3 is indicated. The second panel shows the structure of the initial targeted FKBP5 locus retaining the neo minigene. The primers used to identify the priming sites (3′ PCR_F, 3′ PCR_R) used for Southern blot analysis of ES cells and the AflII and HindIII restriction sites are shown. The bottom panel shows the structure of the final targeted FKBP5 locus after Cre recombination; the locations of the V55L codon substitution in exon 3 and the single remaining LoxP site are shown. The primers used to confirm excision of the neo minigene are also shown (F1, R1). (b) TRAP staining of osteoclasts. BMMs from FKBP51V55L mutant mice and wild-type mice were cultured with varying concentrations of RANKL as indicated and M-CSF (50 ng ml−1) for 3 days. The cells were then stained by TRAP staining, and photomicrographs were obtained using a Nikon Eclipse Ti inverted microscope. Original magnification, × 100. (c) Toluidine blue staining of resorption pits. The photomicrographs were obtained using a Nikon Eclipse Ti inverted microscope. Original magnification, × 100. (d) Numbers of osteoclasts derived from FKBP51V55L mutant and wild-type mice in vitro. When the concentration of RANKL was low (30 ng ml−1), the number of osteoclasts derived from wild-type and FKBP51V55L mutant BMMs did not significantly differ. When the concentration of RANKL was increased (70 ng ml−1, 150 ng ml−1), the number of osteoclasts derived from both wild-type and FKBP51V55L mutant BMMs increased, and the number of osteoclasts derived from FKBP51V55L mutant BMMs was significantly greater than the number obtained from wild-type BMMs. The data shown are representative of three independent experiments. *P<0.05. (e) The areas of resorption pits were calculated using Image Pro Plus. The results show that the mean area of the resorption pits in each field is larger in the mutant group than in the wild-type group. The data shown are representative of three independent experiments. **P<0.01. BMM, bone marrow-derived monocyte/macrophage cells; TRAP, tartrate-resistant acid phosphatase.
The NFACT1 messenger RNA expression level was increased in osteoclast precursors from FKBP51V55L mice; in addition, messenger RNAs for the osteoclast-specific molecules TRAP and the osteoclast-associated receptor were increased in the mutant osteoclast precursors in response to RANKL and M-CSF (Figures 4a–d). Western blotting confirmed that the protein expression level of NFATC1 was increased in osteoclast precursors derived from the mutant mice; however, c-fos expression was not significantly different in the wild-type and mutant groups (Figures 4e and f).

Expression of osteoclast-specific molecules and transcription factors in mice BMMs. (a) Relative expression levels of NFATC1 mRNA. NFACT1 mRNA levels significantly increased following induction by RANKL in both wild-type and FKBP51V55L mutant BMMs. NFATC1 mRNA was higher in FKBP51V55L mutant BMMs than in wild-type BMMs after incubation of the cells with RANKL for 2 days. (b) Relative expression levels of TRAP mRNA. TRAP mRNA expression significantly increased in BMMs after induction by RANKL, and significantly more TRAP was detected in FKBP51V55L mutant BMMs than in wild-type BMMs. (c) Relative expression levels of OSCAR mRNA. OSCAR mRNA expression levels in BMMs were elevated several thousand-fold after induction by RANKL. Significantly more OSCAR mRNA expression was detected in FKBP51V55L mutant BMMs than in wild type. (d) Agarose gel electrophoresis image of one representative result of the real time RT-PCR experiment. (e) One representative western blotting result of c-fos expression in wild-type and FKBP51V55L mutant BMMs in response to RANKL exposure for the indicated time. (f) One representative western blotting result of NFATC1 expression in wild-type and FKBP51V55L mutant BMMs in response to RANKL exposure for the indicated number of days. All data shown are representative of three independent experiments. *P<0.05. OSCAR, osteoclast-associated receptor; TRAP, tartrate-resistant acid phosphatase.
Akt phosphorylation is increased during osteoclast differentiation in osteoclast precursors expressing FKBP51V55L
As shown in Figure 5, Akt phosphorylation in response to RANKL was increased in FKBP51V55L BMMs compared with wild-type BMMs, but no differences in the activation of Erk or the degradation of IκB were found in BMMs of these two genotypes (Figure 5a). Because abnormal autophagy has recently been considered a possible mechanism of PDB pathogenesis, we measured LC3 expression in BMMs during osteoclast differentiation induced by RANKL and M-SCF. The results showed that LC3IIexpression in wild-type and FKBP51V55L BMMs did not significantly differ (Figure 5b).

Detection of osteoclastogenesis-related signaling pathways in mouse BMMs. (a) A representative western blotting result used for signaling pathways detection. Akt phosphorylation is increased in FKBP51V55L mutant BMMs in response to RANKL; however, IκB degradation and ERK phosphorylation show no significant differences in wild-type and mutant BMMs. (b) LC3 expression in BMMs during osteoclast differentiation. LC3II expression in FKBP51V55L mutant and wild-type BMMs induced by RANKL (100 ng ml−1) and M-CSF (50 ng ml−1) is nearly the same. All results are representative of at least two biological replicates.
Intensive trabecular bone resorption phenotype of FKBP51V55L mice
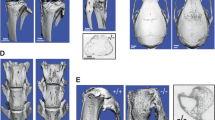
Parametric analysis of the femoral trabecular bone of 10-month-old FKBP51 wild-type and mutant male littermates was performed. This analysis showed that ratio of the bone volume to the total volume of mutant mice was decreased (bone volume ratio FKBP51WT: 0.1401±0.0277; FKBP51V55L: 0.08±0.0219; P=0.0037; trabecular number FKBP51WT: 3.4393±0.5322; FKBP51V55L: 2.2073±0.3871; unit: mm−1; P=0.0020; trabecular thickness FKBP51WT: 0.0406±0.0024; FKBP51V55L: 0.0361±0.0036; unit: mm; P=0.0359). Accordingly, trabecular spacing (FKBP51WT: 0.2561±0.0479, FKBP51V55L: 0.4279±0.0815, unit: mm; P=0.0018) and trabecular pattern factor (FKBP51WT: 20.60±1.50, FKBP51V55L: 25.50±2.61, unit: mm−1; P=0.0035) were increased in the mutant mice (Figure 6a). These results show that there is an intensive trabecular bone resorption pattern in FKBP51V55L mutant mice. However, the average cortical thickness at 1 mm distal to the growth plates of wild-type and mutant mice showed no significant difference (FKBP51WT: 0.1309±0.0111; FKBP51V55L: 0.1385±0.0117; unit: mm; P=0.2982; Figure 6a). Longitudinal micro-CT sections of femurs from wild-type and mutant mice showed that the bone trabeculae of mutant mice were more sparse and thin than were those of the wild-type animals (Figure 6b). No significant cortical thickening or bone expansion was found in the femurs of the mutant animals, and no bending deformity of bone was noted. 3D reconstruction of the distal femurs of two of the three mutant mice showed suspicious osteolytic bone lesions penetrating the cortex; none of the wild-type control animals displayed this phenomenon (Figure 6c).

Micro-CT analysis of femurs from FKBP51WT and FKBP51V55L homozygous mice. (a) Analysis of trabecular bone parameters analysis. Bone volume, trabecular number and tubercular thickness were decreased in FKBP51V55L mice; accordingly, trabecular spacing and trabecular pattern factor increased in FKBP51V55L mice compared with the WT control. However, the average cortical wall thickness in WT and mutant mice shows no significant difference. *P<0.05; **P<0.01. (b) Longitudinal micro-CT sections of femurs from FKBP51WT and FKBP51V55L mutant mice. In the distal femur of the FKBP51V55L mouse, the trabeculae are more sparse and thin than those in the wild-type animal. However, no significant cortical thickening or expansion of the femur from FKBP51V55L mouse is apparent, and no bending deformity of the long bone is seen. (c) Micro-CT analysis with 3D reconstruction of the distal femur and the proximal tibia of a FKBP51WT mouse and of two FKBP51V55L mice. The two mutant mice show suspicious focal osteolytic lesions penetrating the cortex (orange arrows), whereas the same area in the wild-type mouse does not show this phenotype.
Discussion
In many countries, PDB affects ~1–8% of individuals over 55 years of age.21 Individuals with PDB suffer from various bone problems and complications such as bone pain, deformity, fracture, and hearing loss and other nerve root compression symptoms. Genetic factors are important elements in studies of PDB etiology. In this study, we report an extremely rare case of familial PDB found in a northern Han Chinese family. Genetic studies did not reveal mutations in known disease-related genes in this family, and exome sequencing targeted a novel candidate c.163G>C mutation in FKBP5 that is associated with PDB through enhanced osteoclastogenesis. The V55L mutation in FKBP51 was shown to influence the function of FKBP51 in regulating Akt phosphorylation in cells. Osteoclast generation and function studies using FKBP51V55L KI mice showed that the V55L mutation enhanced osteoclastogenesis and osteoclast bone resorption activity in vitro. Akt phosphorylation and NFATC1 expression in BMMs from FKBP51V55L mice were increased compared to normal controls during osteoclast differentiation induced by RANKL. Finally, micro-CT analysis confirmed the presence of intensive trabecular bone resorption in FKBP51V55L mice corresponding to the enhanced osteoclastogenesis noted in vitro, and 3D reconstruction of the distal femurs of the mutant mice showed suspicious bone resorption lesions. These studies indicate that the FKBP5 mutation that we identified is associated with PDB through enhancing osteoclastogenesis.
To date, mutations in SQSTM1 have been widely shown to be associated with PDB through regulation of the RANKL and NF-κB signaling pathways.22 Other susceptibility genes such as TNFRSF11A, TNFRSF11B, OPTN, VCP, CSF1 and DCSAMP have also been reported to be related to or linked to PDB. In our study, with the exception of many SNPs, no mutations in these known susceptibility genes were detected. However, PDB is a rare disease in China. To the best of our knowledge, no more than 200 patients have been reported prior to the study in the literature in English and or in the local language.23, 24 Hence, because they are common, SNPs were not considered likely to be causal, and mutations in unknown genes were further investigated.
Five genetic mutations that co-segregated with patients were identified using whole-exome sequencing in the studied family. One of these five genes, FKBP5, encodes FKBP51, which is involved in the regulation of a number of fundamental biological processes.25 It positively regulates canonical NF-κB activation and negatively modulates Akt via protein–protein interactions.17, 18, 26 Scientists have recently found that FKBP51 also functions in antidepressant therapy by acting as an autophagy inducer.27, 28 Two of the processes that are regulated by FKBP51, NF-κB and Akt signaling, are both involved in osteoclastogenesis. Activation of these two pathways promotes osteoclast development, survival, and bone resorption activation,29, 30, 31, 32, 33, 34 and aberrant NF-κB signaling activation has been confirmed as a critical mechanism in the pathogenesis of PDB. In addition, inefficient autophagy has been presumed to be correlated with the formation of inclusion bodies in the osteoclasts of patients with PDB, and this may be an additional mechanism involved in PDB development. The correlations between the functions of FKBP51 and PDB pathogenesis made the FKBP5 gene a good candidate for studying the disease causative mutations associated with PDB, whereas none of the other four genes in which mutations were found in the affected individuals are known to have functions related to the biological characteristics of osteoclasts.
Functional assays of the mutant FKBP51 protein in cell experiments demonstrated that the V55L mutation in FKBP51 does not affect its regulatory role in NF-κB activation. In contrast, Akt phosphorylation was elevated in HEK293 cells expressing mutant FKBP51, and the kinase activity of Akt was increased in U937 cells expressing mutant FKBP51. It was previously reported that FKBP51 can enhance pAktser473 dephosphorylation by acting as a scaffold for Akt and PHLPP, and that deletion of the FK1 domain (residues 1–138) of FKBP51 abolished its ability to enhance Akt-PHLPP interaction.18 We presume that the FKBP51V55L mutation identified in our study decreases the interaction between Akt and PHLPP, consequently reducing the dephosphorylation of pAktSer473 by PHLPP and ultimately resulting in an increase in Akt phosphorylation. These results emphasize an important role of the V55L mutation in increasing Akt activation, a process that may be involved in osteoclast formation.
Mouse models have been used in studies of the etiology of PDB in attempts to determine whether the known mutations in the SQSTM1 gene or viral proteins can elicit Paget’s-disease-like disorders in mice.10, 35, 36, 37 In the FKBP51V55L KI transgenic mouse model we created here, significantly more osteoclasts were formed from BMMs derived from mutant mice that from control BMMs in response to various concentrations of RANKL. TRAP and osteoclast-associated receptor are two osteoclast-specific molecules that are expressed in osteoclasts during differentiation.38 In vitro experiments showed that both the TRAP and osteoclast-associated receptor messenger RNA levels were significantly increased in FKBP51V55L mutant BMMs compared with wild-type BMMs after induction by RANKL. Pit formation assays revealed that the mean resorption area formed by osteoclasts generated from FKBP51V55L mice was larger than that of the wild type. These results show that osteoclast precursors from FKBP51V55L mice are more sensitive to RANKL than osteoclast precursors from wild-type mice and that the V55L mutation in FKBP51 promotes osteoclast differentiation. In addition, the V55L mutation enhances bone resorption activation in osteoclasts. Taken together, these results strongly suggest that this mutation is related to PDB development by promoting osteoclast differentiation and activation.
NFATC1 is the key transcription factor in osteoclast differentiation, and its induction depends on both the TRAF6 and the c-fos signaling pathways.39 In this study, we found that c-fos expression levels in wild-type and mutant BMMs did not significantly differ; however, NFATC1 levels in FKBP51V55L BMMs were increased during osteoclast differentiation. This indicated that the V55L mutation in FKBP51 directly influences NFATC1 expression independently of c-fos during osteoclastogenesis. Several signaling pathways that are involved in signal transmission during osteoclast differentiation, including the NF-κB, Akt, ERK, JNK and p38 MAPK pathways, depend on the binding of RANKL to its receptor RANK, and these pathways ultimately influence NFATC1 expression.15 We showed that Akt phosphorylation in FKBP51V55L BMMs increased in response to RANKL and that IκB generation and ERK phosphorylation in mutant and wild-type BMMs were not significantly different. This result confirmed the influence of the FKBP51V55L mutation on Akt phosphorylation in mice BMMs and showed that this mutation increases NFATC1 expression and osteoclast differentiation through enhancing Akt activation without influencing the NF-κB and ERK signaling pathways.
Another possible mechanism associated with FKBP51 function that may be involved in PDB development is autophagy. Insufficient autophagy is considered to be related to the formation of inclusion bodies in pagetic osteoclasts. In addition, PDB is a typical age-dependent disease that mainly affects people older than 55 years of age, and aberrant autophagy, which has been linked with aging, may partially explain the typically late onset of PDB.40 FKBP51 has previously been shown to enhance the autophagy process. Our results showed that the expression of LC3II, a biomarker for autophagy, was not significantly different in mutant and wild-type BMMs during the induction of osteoclast differentiation. This indicated that there is no aberrant autophagy in FKBP51V55L mice during osteoclast differentiation in vitro. In summary, the results of our experiments prove that the FKBP51V55L mutation increases osteoclast formation by enhancing the activity of the Akt/NFATC1 pathway in a mouse model in vitro.
The pagetic phenotype has previously been reported in P62P394L transgenic mice. In that study, the authors demonstrated that P62P394L mice develop mixed osteolytic and osteosclerotic bone disorders in the distal femur and proximal tibia with increasing age.37 In our study, the phenotype of the experimental mice was also determined by micro-CT analysis. Excessive trabecular bone resorption was noted in 10-month-old FKBP51V55L mice, corresponding with the enhanced osteoclastogenesis observed in vitro. In addition, resorption bone lesions were detected on a 3D reconstruction of the distal femurs of the mutant mice. The presence of these phenotypes confirmed that the V55L mutation can cause osteolytic bone lesions in vivo. However, osteosclerotic bone disorders involving cortical thickening and expansion were not observed in the mutant mice. This result indicates that the V55L mutation in FKBP51 is insufficient to cause excessive compensatory osteoblastic differentiation in a mouse model. There are two possible reasons for this result: first, the bone micro-environment differs in humans and mice, and these differences may result in different outcomes of osteoblast differentiation in the two species; second, additional gene disorders or environmental factors are needed to trigger aberrant osteoblastic formation in vivo, thereby suggesting an important role of environmental factors in PDB development.
In conclusion, in seeking to identify gene disorders responsible for a Chinese familial PDB, we demonstrated that a novel c.163G>C point mutation in the FKBP5 gene is associated with PDB. The results of our research proved that the FKBP5 mutation, which co-segregated with patients, enhances osteoclast differentiation and bone resorption activity, both of which are primary defects in PDB development. These results indicate that the identified FKBP5 mutation is associated with PDB through enhancing osteoclastogenesis and function. Nevertheless, this work also has the limitation that because none of the mutation carriers in the pedigree are yet over 40 years of age and none of them currently show symptoms of pagetic bone disorder, the available genetic evidence (for example, generation transmission and linkage studies) regarding this mutation is insufficient. Because PDB usually appears at >40 years of age,41 the status of these carriers is now considered unknown, and follow-up visits will be conducted in the future to further study the genetic transmission of this mutation.
References
Takata S, Hashimoto J, Nakatsuka K, Yoshimura N, Yoh K, Ohno I et al. Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24: 359–367.
van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C . Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17: 465–471.
Hashimoto J, Ohno I, Nakatsuka K, Yoshimura N, Takata S, Zamma M et al. Prevalence and clinical features of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24: 186–190.
Seton M, Hansen M, Solomon DH . The implications of the sequestosome 1 mutation P392L in patients with Paget’s disease in a United States cohort. Calcif Tissue Int 2016; 98: 489–496.
Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet 2010; 42: 520–524.
Wuyts W, Van Wesenbeeck L, Morales-Piga A, Ralston S, Hocking L, Vanhoenacker F et al. Evaluation of the role of RANK and OPG genes in Paget’s disease of bone. Bone 2001; 28: 104–107.
Chung PY, Beyens G, de Freitas F, Boonen S, Geusens P, Vanhoenacker F et al. Indications for a genetic association of a VCP polymorphism with the pathogenesis of sporadic Paget’s disease of bone, but not for TNFSF11 (RANKL) and IL-6 polymorphisms. Mol Genet Metab 2011; 103: 287–292.
Chung PY, Beyens G, Boonen S, Papapoulos S, Geusens P, Karperien M et al. The majority of the genetic risk for Paget’s disease of bone is explained by genetic variants close to the CSF1, OPTN, TM7SF4, and TNFRSF11A genes. Hum Genet 2010; 128: 615–626.
Singer FR . Paget’s disease of bone-genetic and environmental factors. Nat Rev Endocrinol 2015; 11: 662–671.
Kurihara N, Hiruma Y, Yamana K, Michou L, Rousseau C, Morissette J et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13: 23–34.
Teramachi J, Nagata Y, Mohammad K, Inagaki Y, Ohata Y, Guise T et al. Measles virus nucleocapsid protein increases osteoblast differentiation in Paget’s disease. J Clin Invest 2016; 126: 1012–1022.
Helfrich MH . Osteoclast diseases. Micros Res Tech 2003; 61: 514–532.
Ralston SH . Pathogenesis of Paget’s disease of bone. Bone 2008; 43: 819–825.
Neale SD, Smith R, Wass JA, Athanasou NA . Osteoclast differentiation from circulating mononuclear precursors in Paget’s disease is hypersensitive to 1,25-dihydroxyvitamin D(3) and RANKL. Bone 2000; 27: 409–416.
Nakashima T, Hayashi M, Takayanagi H . New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab 2012; 23: 582–590.
Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 2004; 6: 97–105.
Romano MF, Avellino R, Petrella A, Bisogni R, Romano S, Venuta S . Rapamycin inhibits doxorubicin-induced NF-kappaB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur J Cancer 2004; 40: 2829–2836.
Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009; 16: 259–266.
Takami M, Kim N, Rho J, Choi Y . Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol 2002; 169: 1516–1523.
Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J . Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc NatI Acad Sci USA 2003; 100: 868–873.
Corral-Gudino L, Borao-Cengotita-Bengoa M, Del Pino-Montes J, Ralston S . Epidemiology of Paget’s disease of bone: a systematic review and meta-analysis of secular changes. Bone 2013; 55: 347–352.
Yip KH, Feng H, Pavlos NJ, Zheng MH, Xu J . p62 ubiquitin binding-associated domain mediated the receptor activator of nuclear factor-kappaB ligand-induced osteoclast formation: a new insight into the pathogenesis of Paget’s disease of bone. Am J Pathol 2006; 169: 503–514.
Gu JM, Zhang ZL, Zhang H, Hu WW, Wang C, Yue H et al. Thirteen Chinese patients with sporadic Paget’s disease of bone: clinical features, SQSTM1 mutation identification, and functional analysis. J Bone Miner Metab 2012; 30: 525–533.
Wat WZ, Cheung WS, Lau TW . A case series of Paget’s disease of bone in Chinese. Hong Kong Med J 2013; 19: 242–248.
Pirkl F, Buchner J . Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. J Mol Biol 2001; 308: 795–806.
Avellino R, Romano S, Parasole R, Bisogni R, Lamberti A, Poggi V et al. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood 2005; 106: 1400–1406.
Gassen NC, Hartmann J, Zschocke J, Stepan J, Hafner K, Zellner A et al. Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: evidence in cells, mice, and humans. PLoS Med 2014; 11: e1001755.
Gassen NC, Hartmann J, Schmidt MV, Rein T . FKBP5/FKBP51 enhances autophagy to synergize with antidepressant action. Autophagy 2015; 11: 578–580.
Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev 1997; 11: 3482–3496.
Jimi E, Nakamura I, Ikebe T, Akiyama S, Takahashi N, Suda T . Activation of NF-kappaB is involved in the survival of osteoclasts promoted by interleukin-1. J Biol Chem 1998; 273: 8799–8805.
Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A et al. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol 2000; 148: 333–342.
Moon JB, Kim JH, Kim K, Youn BU, Ko A, Lee SY et al. Akt induces osteoclast differentiation through regulating the GSK3beta/NFATc1 signaling cascade. J Immunol 2012; 188: 163–169.
Matsumoto T, Nagase Y, Hirose J, Tokuyama N, Yasui T, Kadono Y et al. Regulation of bone resorption and sealing zone formation in osteoclasts occurs through protein kinase B-mediated microtubule stabilization. J Bone Miner Res 2013; 28: 1191–1202.
Cao H, Zhu K, Qiu L, Li S, Niu H, Hao M et al. Critical role of AKT protein in myeloma-induced osteoclast formation and osteolysis. J Biol Chem 2013; 288: 30399–30410.
Kurihara N, Hiruma Y, Zhou H, Subler MA, Dempster DW, Singer FR et al. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest 2007; 117: 133–142.
Hiruma Y, Kurihara N, Subler MA, Zhou H, Boykin CS, Zhang H et al. A SQSTM1/p62 mutation linked to Paget’s disease increases the osteoclastogenic potential of the bone microenvironment. Hum Mol Genet 2008; 17: 3708–3719.
Daroszewska A, van’t Hof RJ, Rojas JA, Layfield R, Landao-Basonga E, Rose L et al. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget’s disease-like disorder in mice. Hum Mol Genet 2011; 20: 2734–2744.
Kim K, Kim JH, Lee J, Jin HM, Kook H, Kim KK et al. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 2007; 109: 3253–3259.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002; 3: 889–901.
Chung PY, Van Hul W . Paget’s disease of bone: evidence for complex pathogenetic interactions. Semin Arthritis Rheum 2012; 41: 619–641.
Klein RM, Norman A . Diagnostic procedures for Paget’s disease. Radiologic, pathologic, and laboratory testing. Endocrinol Metab Clin North Am 1995; 24: 437–450.
Acknowledgements
We thank all participants involved in this study. This work was supported by the National Basic Research Program of China (973 program-2012CB944703), the National Natural Science Foundation of China (Grant No 31370897) and the Shandong Provincial Natural Science Foundation (Grant No ZR2012HM054).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lu, B., Jiao, Y., Wang, Y. et al. A FKBP5 mutation is associated with Paget’s disease of bone and enhances osteoclastogenesis. Exp Mol Med 49, e336 (2017). https://doi.org/10.1038/emm.2017.64
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.64
This article is cited by
-
Comparative Analysis of Differentially Expressed Genes in Chondrocytes from Rats Exposed to Low Selenium and T-2 Toxin
Biological Trace Element Research (2024)
-
Paget’s Disease of Bone
Calcified Tissue International (2019)
-
Human Genetics of Sclerosing Bone Disorders
Current Osteoporosis Reports (2018)
