
Abstract
The Raf kinase inhibitory protein (RKIP) has been demonstrated to modulate different intracellular signaling pathways in cancers. Studies have shown that RKIP is frequently downregulated in cancers; therefore, attempts have been made to upregulate the expression of RKIP using natural and synthetic agents for the treatment of human malignancies. Moreover, various regulators such as specific proteins and microRNAs (miRNAs) that are involved in the regulation of RKIP expression have also been identified. RKIP mechanistically modulates the apoptotic regulators of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling. Because of its critical role in human cancers, RKIP has drawn much research attention, and our understanding is expanding rapidly. Here, we summarize some of the biological complexities of RKIP regulation. However, we restrict our discussion to selected tumors by focusing on TRAIL, miRNAs and natural agents. Emerging evidence suggests a role for natural agents in RKIP regulation in cancer cells; therefore, naturally occurring agents may serve as cancer-targeting agents for cancer treatment. Although the literature suggests some advancement in our knowledge of RKIP biology, it is incomplete with regard to its preclinical and clinical efficacy; thus, further research is warranted. Furthermore, the mechanism by which chemotherapeutic drugs and novel compounds modulate RKIP and how nanotechnologically delivered RKIP can be therapeutically exploited remain to be determined.
Similar content being viewed by others

Introduction
Raf (rapidly accelerated fibrosarcoma1) kinases are a family of three serine/threonine-specific protein kinases that are related to retroviral oncogenes.2 For example, mouse sarcoma virus 3611 contains a Raf kinase-related oncogene that enhances fibrosarcoma induction. RAF kinase activity is regulated by its endogenous inhibitor Raf kinase inhibitory protein (RKIP). The well-studied tumor suppressor RKIP belongs to a family of evolutionarily conserved phosphatidylethanolamine-binding proteins. Detailed mechanistic insights have provided compelling evidence of the critical role of RKIP in the modulation of different signaling cascades (discussed below). RKIP inhibits MAP kinase (MAPK) signaling because RKIP-bound Raf‐1 is unable to undergo phosphorylation by its activator. Indeed, protein kinase C-mediated phosphorylation of RKIP at S153 sequesters it away from Raf-1, thereby allowing Raf-1 to transduce signals to downstream effectors.3 Surprisingly, phospho-RKIP exerted inhibitory effects on G protein‐coupled receptor kinase‐2 (GRK‐2), which is a feedback inhibitor for coupled receptor.4 RKIP is frequently inactivated during cancer progression, as evidenced by the methylated RKIP promoter in gastric adenocarcinoma tissues. RKIP expression has been found to be correlated with Union for International Cancer Control (UICC) stage, pathological stage and lymph node metastasis. Significantly lower five-year overall survival rates were noted in patients with RKIP− tumors as compared with RKIP+ patients.5 Below we elaborate on the role of RKIP in selected human cancers.
The role of RKIP in prostate cancer
Prostate cancer (PCa) is a genomically complex disease. Recently emerging scientific reports have confirmed that androgen-independent cancer progression is a scientific and clinical challenge. Nearly all PCa patients treated with androgen ablation therapy eventually develop castrate-resistant PCa, which subsequently metastasizes; there is no cure for the metastatic disease. Metastasis is a complex and multistep biological mechanism consisting of several hallmark features, including epithelial–mesenchymal transition, invasion, extravasation from vessels, intravasation into lymph vessels and blood and colonization via the metastatic ability of competent cancer cells. During this process, matrix metalloproteinases (MMPs) efficiently degrade the extracellular matrix (ECM) and basement membrane to generate space for cellular dissemination from the original site. Tissue inhibitor of metalloproteinases exerts inhibitory effects on MMPs, although the role of tissue inhibitor of metalloproteinases is highly complex in metastatic disease. Transgenic adenocarcinoma of the mouse prostate (TRAMP) has been developed to closely study the molecular mechanisms underlying human PCa.
Recently, progressing tumors in TRAMP (+) mice have been convincingly revealed to endogenously express considerably decreased levels of RKIP. Primary tumors were found to develop earlier in RKIP (−/−) TRAMP (+) mice as compared with TRAMP (+) mice. Moreover, distant metastasis was evident in the RKIP (−/−) TRAMP (+) mice. Prostate epithelial cells proliferated at a higher rate in the RKIP (−/−) TRAMP (+) mice in the 10th and 20th weeks as compared with the TRAMP (+) mice.6 Photodynamic therapy, a clinically approved treatment reported to effectively target PCa cells, has been shown to trigger nitric oxide generation by upregulating nitric oxide synthases. There is evidence of cytoprotective effects of nitric oxide in PCa cells non-optimally treated with photodynamic therapy.7
Microtubule inhibitors have been found to enhance RKIP expression in DU145 cells. It was noted that DU145 cells transfected with RKIP exhibited a greater than threefold increase in apoptosis; the results were pronounced upon treatment of RKIP-overexpressing PCa cells with microtubule inhibitors.8 The androgen receptor has been experimentally verified to bind to response elements in the RKIP promoter; as expected, anti-androgen bicalutamide inhibited androgen receptor-mediated transcriptional regulation of RKIP.9 RKIP has also been reported to have a significant role in breast cancer. The following section describes strategies to stimulate the expression of RKIP in cultured breast cancer cells. These strategies could be fully exploited in the future for the treatment of breast cancer and other human cancers.
The role of RKIP in breast cancer
The biological features of RKIP-expressing triple-negative breast cancer cells have been studied, and transfection assays revealed that expression levels of MMP-1 and MMP-2 were markedly reduced in RKIP-expressing MDA-MB-231 cells.10
Gemifloxacin is a fluoroquinolone antibiotic that reportedly inhibits the migratory and invasive potential of MDA-MB-453 and MDA-MB-231 breast cancer cells. Phosphorylation of IκB and functionally active NFKB was inhibited in gemifloxacin-treated breast cancer cells.11 4-Shogaol, a phytochemical extracted from red ginger, has been shown to exert inhibitory effects on phosphorylation of IκB and NF-kB in breast cancer cells. RKIP was demonstrated to be increased in 4-Shogaol-treated breast cancer cells, and depletion of RKIP impaired 4-Shogaol-mediated biological effects. Tumor growth was also remarkably reduced in xenografted mice treated with 4-Shogaol.12 Next, we discuss how RKIP interacts with other proteins to transduce signals in cancer cells.
The crosstalk of RKIP with other proteins
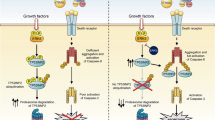
Structural studies have shown that RKIP physically interacts with MDA-9 to impair its association with Src, which is summarized in a diagram (Figure 1). The MDA-9–Src interaction occurs through PDZ domains and consequently enhances formation of a signalosome consisting of stable c-Src/focal adhesion kinase (FAK) in order to transduce signals to downstream effectors.13 Metastatic melanoma cells grown on fibronectin-coated plates display higher levels of MDA-9 and increased Src phosphorylation at Tyr418 and FAK phosphorylation at Tyr397. An MDA-9-mediated increase in the phosphorylated states of Src and FAK was shown to be dramatically reduced in RKIP-overexpressing cancer cells.13

MDA-9-mediated regulation of activation of Src and FAK. RKIP negatively regulates MDA-9.
However, in another report, apoptosis and the cell proliferation rate were not influenced by RKIP overexpression or RKIP silencing in cholangiocarcinoma RBE cells. RKIP-expressing cells had reduced MMP levels and upregulated tissue inhibitor of metalloproteinases levels.14 Likewise, RKIP considerably inhibited the invasive and metastasizing potential of esophageal cancer cells via the downregulation of MMP14.15 Increased expression of RKIP has also been reported to inhibit the distantly metastasizing potential of advanced nasopharyngeal carcinoma cells.16 Immunohistochemistry has shown that RKIP expression is predictive of uveal melanoma metastasis.17 Moreover, in addition to the Raf kinase, the FAK was autophosphorylated at Tyr397, consequently generating a binding site for the Src-homology (SH)2 domain of Src. Src-induced FAK phosphorylation at Tyr576 and Tyr577 further enhanced the catalytic activity of FAK.18 This finding also generated intense interest and resulted in the development of several Src inhibitors for cancer treatment.
In the following section, we discuss how RKIP overcomes resistance against TRAIL-induced apoptosis in resistant cancer cells. The emphasis is on two important oncogenic proteins—Yin Yang 1 (YY1) and Snail—and on strategies that can be utilized to improve the apoptotic rate in resistant phenotype cells.
Positive regulation of TRAIL-induced apoptosis by RKIP
Data obtained through high-throughput technologies have provided compelling evidence supporting the role of RKIP as a tumor suppressor. Moreover, RKIP modulates intracellular signaling networks to overcome resistance against molecular therapeutics. Apoptosis is characteristically triggered primarily through two extensively studied pathways: the extrinsic and intrinsic apoptotic pathways. The extrinsic pathway is functionalized as a result of the interaction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), TNFα and FasL with their receptors (TRAIL receptor, TNFR and Fas, respectively). A signalosome is formed at a death receptor consisting of FADD and pro-caspase-8 (Figure 2a). Activated caspase-8 activates its downstream effector caspase-3. By contrast, the intrinsic pathway is activated through the entry of caspase-8-mediated proteolytically processed Bid into the mitochondrion to increase the permeability of the mitochondrial membrane. Then, Cytosolically accumulated cytochrome c interacts with APAF and pro-caspase-9 to form the apoptosome.

(a) TRAIL-induced signalosome formation at the death receptor consisting of FADD and Caspase-8. RKIP enhances TRAIL-induced apoptosis by upregulating death receptors. (b) NF-κB activates Snail, which further inhibits RKIP. Inhibitor of Kappa B (IκB) inactivates NF-κB, and therefore SNAIL is inhibited. NPI-0052 has been shown to enhance IκB levels. LFB-R603 inhibits NF-κB and Snail. Snail is also inhibited by galiximab. (c) The YY1 protein transcriptionally downregulates DR5, whereas RKIP and galiximab inhibit the YY1 protein.
TRAIL-mediated signaling has emerged as one of the most deeply studied molecular mechanisms reported to induce apoptosis in cancer cells while leaving normal cells intact. Preclinical and clinical studies have greatly enhanced our understanding of TRAIL-induced intracellular signaling through a death receptor (for example, DR4 and DR5). There is a stoichiometric ratio of pro-apoptotic and anti-apoptotic proteins, and imbalance of these proteins considerably impairs TRAIL-induced apoptosis. Moreover, downregulation of DR4 or DR5 and overexpression of either cFLIP (negative regulator) or NFKP can also negatively regulate apoptotic cell death. In the following section, we discuss how RKIP positively regulates TRAIL-induced apoptosis by efficiently enhancing DR5 expression in cancer cells and the different strategies that have been used to induce RKIP expression in cancer cells. These findings are summarized in Figures 2a–c.
YY1 is a transcriptional repressor with Fas and DR5 promoter regions contained in its binding sites (Figure 2c). Thus, the transcriptional downregulation of DR5 will impair TRAIL-induced apoptosis in cancer cells. RKIP negatively regulatesYY1 expression, as evidenced by the decrease in YY1 promoter activity in RKIP-overexpressing cancer cells.19 Treatment of RKIP-overexpressing PCa cells with increasing concentrations of TRAIL resulted in a markedly enhanced apoptotic rate;19 however, RKIP-silenced cells did not respond to TRAIL and a chemotherapeutic drug. DR5 promoter activity was significantly enhanced in RKIP-overexpressing PCa cells transfected with a wild-type DR5 reporter construct,19 whereas YY1 promoter activity was considerably reduced in PCa cells transfected with an RKIP expression vector. Moreover, procaspase 8 activation was enhanced in RKIP-overexpressing PCa cells following treatment with TRAIL.19 Mechanistically, constitutively high basal levels of c-FLIPL were shown to be considerably reduced in RKIP-overexpressing PCa cells. Bcl-xL and XIAP levels were also significantly downregulated in RKIP-overexpressing cancer cells.19
Snail is a transcriptional repressor of RKIP that is partially positively regulated by NF-κB in cancer cells. Therefore, cancer cells with functionally active NF-kB show considerably reduced RKIP levels. NPI-0052 (salinosporamide A), a proteasome inhibitor family member isolated from marine Salinisporatropica, has been shown to effectively inhibit NF-κB promoter activity in DU-145 cells. Phosphorylated IκBα levels were also markedly enhanced in NPI-0052-treated cancer cells. Treatment of cancer cells with the NF-κB inhibitor DHMEQ inhibited Snail mRNA expression and increased RKIP transcript levels in a time-dependent manner.20 A similar approach was used to design an effectively designed chimeric monoclonal anti-CD20 mouse/human antibody (LFB-R603) with a human IgG1 constant (Fc) region; this antibody was reported to significantly inhibit the ability of NF-kB and Snail to upregulate the expression of RKIP in non-Hodgkin B-cell lymphoma (Figure 2b).21 Thus, NPI-0052 and LFB-R603 have been shown to overcome resistance against TRAIL in cancer cells.
NF-κB was previously reported to trigger the expression of Snail and YY1 in cancer cells.22, 23 Galiximab (IDEC-11), a high-affinity primatized monoclonal antibody, considerably improved the synergistic effects of TRAIL and cisplatin in a Burkitt B-NHL (non-Hodgkin lymphoma) cell line. Both YY1 and Snail were inhibited in galiximab-treated cancer cells (Figure 2b). Additionally Bcl-xL was also regulated by YY1 and Snail, as evidenced by the downregulated expression of Bcl-xL in YY1- and Snail-inhibited cancer cells.24 TRAIL-mediated differential killing of cancer cells has been recommended for entry into different phases of clinical trials. Although it exhibited notable clinical efficacy in different cancer patients, some associated off-target effects and resistance against TRAIL have been reported. RKIP’s role in improving TRAIL-induced apoptosis has been insufficiently studied in preclinical studies. This role needs to be investigated, including the mechanism by which RKIP re-balances pro- and anti-apoptotic proteins in cancer cells to overcome resistance against TRAIL. In addition, future studies must converge on the identification of natural agents with remarkable potential to upregulate RKIP; this strategy could be used to maximize TRAIL-induced apoptosis in cancer cells.
In the following section, we discuss what is known about the role of miRNA in the regulation of RKIP. miRNA biology has undergone substantial broadening, and emerging evidence has begun to scratch the surface of a new interplay between miRNA and RKIP. Thus, the following section mainly addresses miRNA-mediated control of RKIP in cancer cells.
The role of microRNAs (miRNAs) and their interplay with RKIP
miRNAs are non-coding RNA molecules approximately 22 nucleotides long that are characteristically categorized as tumor suppressor and oncogenic miRNAs. Research over the years has sequentially unveiled considerable contributions by these miRNAs in the modulation of multiple biological mechanisms that were once considered ‘genomic trash.’
A549/CDDP lung adenocarcinoma cells are cisplatin-resistant and exhibit upregulated expression of miR-27a. RKIP is post-transcriptionally controlled by miR-27a (Table 1), and downregulation of RKIP impairs chemotherapeutic drug-induced apoptotic responses in cancer cells.25 Knockdown of RKIP resulted in an increase in vimentin and reduction in E-cadherin levels,25 suggesting a regulatory role for RKIP at the epithelial–mesenchymal transition. Therefore, silencing of miR-27a using antisense oligonucleotides appears to be essential for restoring the drug-induced apoptotic response in drug-resistant cancer cells.
RKIP expression has been found to be significantly reduced in SGC-7901 cells transfected with an miR-224 mimic, thus confirming that miR-224 quantitatively regulates RKIP in gastric cancer SGC-7901 cells.26 The miR-224 is also frequently overexpressed in hepatocellular carcinoma Hep3B cells.27 MDA-MB-231, a highly invasive breast cancer cell line, exhibited upregulated expression of miR-224. These results suggest a role for miR-224 in the regulation of RKIP (Table 1).
LIN28, HMGA2, SDC2 and LOX: RKIP leads microwarriors from the frontline
LIN28 is a frequently overexpressed and well-studied inhibitor of let-7 miRNA biogenesis. LIN28 binds to the pre-element of pri- or pre-let-7, rendering it unable to undergo Drosha- or Dicer-mediated processing. In addition, LIN28-mediated 3′-end oligouridylation of pre-let-7 also inhibits Dicer-mediated molecular processing.28 Interestingly, Lin28 is negatively regulated by RKIP in cancer cells.29
The high-mobility group AT-hook 2 (HMGA2), Syndecan-2 (SDC2) and Lysyl Oxidase (LOX) have previously been documented to be instrumental in metastasis. There is evidence of HMGA2-mediated inhibition of the tumor suppressor miR-200b in cancer cells. However, miR-200b was significantly enhanced in RKIP-overexpressing cancer cells (Table 1). It was evident from the present study that RKIP inhibited the expression of SDC2 and LOX, thereby inhibiting the metastasizing ability of cancer cells.30 Both the primary miR-200b transcript and the mature RNA were notably expressed in HMGA2-depleted 1833 cells; additionally, HMGA2-depleted 1833 cells exhibited reduced LOX expression.30 MDA-MB-436 and MDA-MB-231 cells are ER− breast cancer cells; transfecting this cell lines with the let-7g precursor led to the upregulation of miR-200b and consequently inhibited the expression of LOX and HMGA2.30 There was a negative correlation between Syndecan-2 (SDC2) and RKIP, as evidenced by the markedly enhanced SDC2 expression in RKIP-depleted MDA-MB-435 cells. Tumor growth was also considerably inhibited in mice xenografted with SDC2-silenced 1833 cells.30 These results suggest that RKIP’s multi-targeted approach can be efficiently exploited to inhibit cancer progression. It will be even more useful to develop a broader picture of miRNAs that are under the direct control of RKIP.
It has been experimentally verified in let‐7a-overexpressing MCF10A cells that gene silencing of RKIP dramatically reduces let‐7a and let‐7g expression.29 LIN28 was reported to be a negative regulator of let‐7, because RKIP induced the reversion of let‐7a and let‐7g back to basal levels in LIN28-expressing cells.29 RKIP suppressed the invasive and metastasizing potential of cancer cells by inhibiting LIN28, and LIN28-silenced cancer cells mimicked RKIP-overexpressing cancer cells. For instance, bone metastasis was almost completely inhibited in mice injected in the cardiac ventricle with LIN28‐depleted cells.29
miR-98 was found to be negatively correlated with HMGA2 mRNA levels and positively correlated with RKIP mRNA levels in glioma tissues and cell lines (Table 1). U251 and U87 cells co-transfected with RKIP and miR-98 mimics demonstrated a robust expression of miR-98 along with considerable miR-98-mediated inhibition of HMGA2 protein expression. Moreover, RKIP- and miR-98-expressing U251 and U87 cells exhibited remarkably reduced invasive potential.31 These results suggest that exogenously supplied RKIP and miR-98 work synergistically to inhibit the metastasizing potential of cancer cells.
Thus, a hierarchically organized network seems to contribute to cancer progression. The information summarized in this section provides insights into a tightly regulated protein network consisting of LIN28, HMGA2, SDC2 and LOX that coordinates to induce cancer progression. However, RKIP exerts inhibitory effects on these proteins by upregulating the expression of tumor suppressor miRNAs.
Interplay of BACH1 and RKIP
BACH1, a leucine zipper transcriptional factor involved in metastasis, was markedly reduced in wild-type RKIP-expressing cancer cells. It is reportedly regulated by let‐7, which has been linked to Lin28. RKIP induced let‐7 and negatively regulated BACH1, resulting in reduced expression of its target genes, CXCR4 and MMP1.32 These findings suggest that the regulation of RKIP is a complex process involving multiple genes and miRNAs, and therefore novel strategies by which RKIP could be induced in tumors will represent a breakthrough in cancer treatment. The following section discusses the known mechanism underlying RKIP regulation.
The mechanism underlying RKIP regulation
RKIP expression is controlled by various proteins. In the following section, we list the proteins reported to regulate RKIP expression. The list includes EZH2 and BACH, thus shedding light on the mechanisms through which RKIP expression is controlled.
Recruitment of the suppressor of zeste 12 (Suz12) and EZH2 to the proximally located E-boxes of the RKIP promoter resulted in H3-K9-me3 and H3-K27-me3 modifications. Mechanistically, the SET domain was shown to be mutated in EZH2-expressing MCF-7 cells and did not reduce RKIP expression levels. The methyltransferase activity of the mutant EZH2 was impaired, thus, RKIP expression remained unchanged. However, wild-type EZH2-expressing cancer cells exhibited markedly reduced RKIP levels. Interestingly, miR-101 negatively regulated EZH2, and PCa DU145 cells ectopically expressing miR-101 showed increased RKIP expression.33 These results suggest that members of the epigenetic-modifying protein machinery can be targeted through miRNA. There is a need to identify natural agents that are non-toxic to humans in general with notable epigenetic-modifying ability in order to enhance RKIP expression in cancer cells. Such agents may be useful for cancer prevention and therapy through the upregulation of RKIP.
Astonishingly, it has been shown that BACH1 negatively regulates the expression of RKIP in triple negative breast cancer cells, as evidenced by twofold rise in RKIP protein and mRNA expression in BACH1-depleted MDA-MB-231 cells. BACH1 was reported to bind to a −3000 bp region located upstream of the RKIP transcriptional start site. Interestingly, BACH1 was also involved in autoregulation, as verified by a downstream region (+1000 bp) of the BACH1 promoter that contained a BACH1 binding sequence. Mechanistically, the repression of RKIP was shown to be an orchestrated mechanism in which prior binding of BACH1 to the RKIP promoter had a significant role in facilitating the binding of EZH2 at the promoter site.34 The question remains as to how RKIP could be modulated by novel strategies. The identification of natural agents for exploitation is described in the following section.
Natural agents mediated regulation of RKIP in cancer cells
Based on the existing literature, RKIP appears to be a target for cancer therapy (as documented above) that could be easily achievable using natural agents (the naturopathic approach). Modulation of RKIP using different natural agents is gaining increasing attention. This section examines evidence related to herbal agents reported to modulate RKIP expression in cancer cells.
RKIP expression was considerably enhanced in pancreatic adenocarcinoma cells treated with epigallocatechin gallate (EGCG). Additionally, EGCG treatment exerted inhibitory effects on both the nuclear accumulation of NF-kB and functionally active ERK.35 Dihydroartemisinin remarkably upregulated RKIP expression in HeLa cells; moreover, tumor growth was reduced in xenografted mice treated with dihydroartemisinin.36 The flavonoid didymin also enhanced RKIP expression in neuroblastoma cells, and tumor growth was considerably inhibited in xenografted mice treated with didymin.37 American ginseng (Panaxquinquefolius L.) extract has been shown to reduce phospho-ERK1/2 and -MEK1/2 levels and increase RKIP and phospho-Raf-1 in breast cancer cells.38 These limited studies provide incentives for further investigations to elucidate the role of naturopathic approaches in the upregulation of RKIP, which could be useful for designing future studies in human cancers. Moreover, it is our opinion that chemicals that exist in nature must be harnessed to exploit the identification of specific agents that could be useful for RKIP upregulation. This approach will contribute to the future of personalized molecularly targeted cancer therapy.
Conclusion and perspectives
The topic of RKIP biology has undergone substantial expansion over the years. RKIP is now known to be an effective tumor suppressor that is frequently downregulated in cancer. Nanotechnological delivery of RKIP requires further research because we have no evidence related to the targeting of cancer cells using nanoparticle-conjugated RKIP genes. The use of natural agents is a deeply studied area; the characterization of potential candidates with potent anticancer activity that may induce the expression of RKIP is required. There is evidence of upregulation of RKIP by natural agents, but detailed analyses of transcriptional and protein networks will add sufficient evidence related to their efficacy. Moreover, the mechanism by which RKIP modulates the balance between pro- and anti-apoptotic proteins in drug-resistant and TRAIL-resistant cancer cells requires further investigation. Because RKIP inhibits many kinases, the development of a broader picture of the Kinome that comes directly under RKIP regulation is needed. An improved understanding of the molecular biology of RKIP and its regulation by miRNAs will open new avenues for the development of clinically effective therapeutics that will likely revolutionize cancer treatment.
References
Zebisch A, Troppmair J . Back to the roots: the remarkable RAF oncogene story. Cell Mol Life Sci 2006; 63: 1314–1330.
Roskoski R Jr . RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun 2010; 399: 313–317.
Kolch W . Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 2005; 6: 827–837.
Lorenz K, Lohse MJ, Quitterer U . Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003; 426: 574–579.
Li DX, Cai HY, Wang X, Feng YL, Cai SW . Promoter methylation of Raf kinase inhibitory protein: a significant prognostic indicator for patients with gastric adenocarcinoma. Exp Ther Med 2014; 8: 844–850.
Escara-Wilke J, Keller JM, Ignatoski KM, Dai J, Shelley G, Mizokami A et al. Raf kinase inhibitor protein (RKIP) deficiency decreases latency of tumorigenesis and increases metastasis in a murine genetic model of prostate cancer. Prostate 2015; 75: 292–302.
Della PE, Simonella F, Bonavida B, Xodo LE, Rapozzi V . Repeated sub-optimal photodynamic treatments with pheophorbide a induce an epithelial mesenchymal transition in prostate cancer cells via nitric oxide. Nitric Oxide 2015; 45: 43–53.
Yousuf S, Duan M, Moen EL, Cross-Knorr S, Brilliant K, Bonavida B et al. Raf kinase inhibitor protein (RKIP) blocks signal transducer and activator of transcription 3 (STAT3) activation in breast and prostate cancer. PLoS One 2014; 9: e92478.
Zhang H, Wu J, Keller JM, Yeung K, Keller ET, Fu Z . Transcriptional regulation of RKIP expression by androgen in prostate cells. Cell Physiol Biochem 2012; 30: 1340–1350.
Hao C, Wei S, Tong Z, Li S, Shi Y, Wang X et al. The effects of RKIP gene expression on the biological characteristics of human triple-negative breast cancer cells in vitro. Tumour Biol 2012; 33: 1159–1167.
Chen TC, Hsu YL, Tsai YC, Chang YW, Kuo PL, Chen YH . Gemifloxacin inhibits migration and invasion and induces mesenchymal-epithelial transition in human breast adenocarcinoma cells. J Mol Med (Berl) 2014; 92: 53–64.
Hsu YL, Chen CY, Lin IP, Tsai EM, Kuo PL, Hou MF . 4-Shogaol, an active constituent of dietary ginger, inhibits metastasis of MDA-MB-231 human breast adenocarcinoma cells by decreasing the repression of NF-kappaB/Snail on RKIP. J Agric Food Chem 2012; 60: 852–861.
Das SK, Bhutia SK, Sokhi UK, Azab B, Su ZZ, Boukerche H et al. Raf kinase inhibitor RKIP inhibits MDA-9/syntenin-mediated metastasis in melanoma. Cancer Res 2012; 72: 6217–6226.
Ma J, Shi J, Zhao D, Cheng L, Wang W, Li F et al. Raf kinase inhibitor protein inhibits cholangiocarcinoma cell metastasis by downregulating matrix metalloproteinase 9 and upregulating tissue inhibitor of metalloproteinase 4 expression. Oncol Lett 2015; 9: 15–24.
Zhao D, Ma J, Shi J, Cheng L, Li F, Jiang X et al. Raf kinase inhibitor protein inhibits esophageal cancer cell invasion through downregulation of matrix metalloproteinase expression. Oncol Rep 2013; 30: 304–312.
Li SW, Wang H, Liu ML, Zhang HB, Xiang YQ, Lv X et al. Positive effect of high RKIP expression on reduced distant metastasis by chemotherapy when combined with radiotherapy in locoregionally advanced nasopharyngeal carcinoma: a prospective study. Med Oncol 2013; 30: 322.
Caltabiano R, Puzzo L, Barresi V, Cardile V, Loreto C, Ragusa M et al. Expression of Raf kinase inhibitor protein (RKIP) is a predictor of uveal melanoma metastasis. Histol Histopathol 2014; 29: 1325–1334.
Mitra SK, Hanson DA, Schlaepfer DD . Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005; 6: 56–68.
Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B . Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J Immunol 2007; 179: 5441–5453.
Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B . Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res 2009; 69: 8376–8385.
Baritaki S, Militello L, Malaponte G, Spandidos DA, Salcedo M, Bonavida B . The anti-CD20 mAb LFB-R603 interrupts the dysregulated NF-kappaB/Snail/RKIP/PTEN resistance loop in B-NHL cells: role in sensitization to TRAIL apoptosis. Int J Oncol 2011; 38: 1683–1694.
Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van RF et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007; 26: 7445–7456.
Bonavida B, Baritaki S . Dual role of NO donors in the reversal of tumor cell resistance and EMT: downregulation of the NF-kappaB/Snail/YY1/RKIP circuitry. Nitric Oxide 2011; 24: 1–7.
Martinez-Paniagua MA, Vega MI, Huerta-Yepez S, Baritaki S, Vega GG, Hariharan K et al. Galiximab signals B-NHL cells and inhibits the activities of NF-kappaB-induced YY1- and snail-resistant factors: mechanism of sensitization to apoptosis by chemoimmunotherapeutic drugs. Mol Cancer Ther 2012; 11: 572–581.
Li J, Wang Y, Song Y, Fu Z, Yu W . miR-27a regulates cisplatin resistance and metastasis by targeting RKIP in human lung adenocarcinoma cells. Mol Cancer 2014; 13: 193.
Liu H, Li P, Li B, Sun P, Zhang J, Wang B et al. RKIP suppresses gastric cancer cell proliferation and invasion and enhances apoptosis regulated by microRNA-224. Tumour Biol 2014; 35: 10095–10103.
Poma P, Labbozzetta M, Vivona N, Porcasi R, D'Alessandro N, Notarbartolo M . Analysis of possible mechanisms accounting for raf-1 kinase inhibitor protein downregulation in hepatocellular carcinoma. OMICS 2012; 16: 579–588.
Mayr F, Heinemann U . Mechanisms of Lin28-mediated miRNA and mRNA regulation—a structural and functional perspective. Int J Mol Sci 2013; 14: 16532–16553.
Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM et al. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J 2009; 28: 347–358.
Sun M, Gomes S, Chen P, Frankenberger CA, Sankarasharma D, Chung CH et al. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene 2014; 33: 3528–3537.
Chen Z, Cheng Q, Ma Z, Xi H, Peng R, Jiang B . Overexpression of RKIP inhibits cell invasion in glioma cell lines through upregulation of miR-98. Biomed Res Int 2013; 2013: 695179.
Yun J, Frankenberger CA, Kuo WL, Boelens MC, Eves EM, Cheng N et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J 2011; 30: 4500–4514.
Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res 2012; 72: 3091–3104.
Lee J, Lee J, Farquhar KS, Yun J, Frankenberger CA, Bevilacqua E et al. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc Natl Acad Sci U S A 2014; 111: E364–E373.
Kim SO, Kim MR . (-)-Epigallocatechin 3-gallate inhibits invasion by inducing the expression of Raf kinase inhibitor protein in AsPC1 human pancreatic adenocarcinoma cells through the modulation of histone deacetylase activity. Int J Oncol 2013; 42: 349–358.
Hu CJ, Zhou L, Cai Y . Dihydroartemisinin induces apoptosis of cervical cancer cells via upregulation of RKIP and downregulation of bcl-2. Cancer Biol Ther 2014; 15: 279–288.
Singhal J, Nagaprashantha LD, Vatsyayan R, Ashutosh, Awasthi S, Singhal SS . Didymin induces apoptosis by inhibiting N-Myc and upregulating RKIP in neuroblastoma. Cancer Prev Res (Phila) 2012; 5: 473–483.
King ML, Murphy LL . American ginseng (Panax quinquefolius L.) extract alters mitogen-activated protein kinase cell signaling and inhibits proliferation of MCF-7 cells. J Exp Ther Oncol 2007; 6: 147–155.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Farooqi, A., Li, Y. & Sarkar, F. The biological complexity of RKIP signaling in human cancers. Exp Mol Med 47, e185 (2015). https://doi.org/10.1038/emm.2015.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2015.70
This article is cited by
-
Overexpression of RKIP and its cross-talk with several regulatory gene products in multiple myeloma
Journal of Experimental & Clinical Cancer Research (2017)
