
Abstract
Many researchers have reported that oxidative damage to mitochondrial DNA (mtDNA) is increased in several age-related disorders. Damage to mitochondrial constituents and mtDNA can generate additional mitochondrial dysfunction that may result in greater reactive oxygen species production, triggering a circular chain of events. However, the mechanisms underlying this vicious cycle have yet to be fully investigated. In this review, we summarize the relationship of oxidative stress-induced mitochondrial dysfunction with mtDNA mutation in neurodegenerative disorders.
Similar content being viewed by others
Introduction
Numerous lines of evidence indicate that mitochondrial dysfunction is related to the aging process, and also to neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease and amyotrophic lateral sclerosis.1, 2, 3 Abnormal production of oxidative stress and excessive accumulation of mitochondrial DNA (mtDNA) mutations result in mitochondrial dysregulation, a direct cause of aging4 (Table 1). However, the direct relationship between mtDNA mutation and the generation of reactive oxygen species (ROS) is still questionable. Because mtDNA repair enzymes are limited in number, and mtDNA is easily affected by ROS generation, it is more vulnerable to oxidative stress than nuclear DNA.5, 6 Furthermore, the accumulation of mtDNA mutations could decrease the capability of the electron transport chain, triggering decreased adenosine triphosphate production and increased ROS production. Conversely, increased ROS generation could result in the accumulation of further mtDNA mutations, establishing a feedback loop of mtDNA mutation and ROS generation that contributes to cell death.7, 8 In this review, we provide an update on the relationship between oxidative stress-induced mtDNA mutation and cellular homeostasis.
Basic mitochondrial genetics
Mitochondrial ROS and aging
Mitochondria are believed to contribute to aging through the accumulation of mtDNA mutations and the generation of reactive oxygen species.9 The conventional view is that mitochondria regulate cellular homeostasis by producing several redox enzymes, but excessive generation of ROS impairs mitochondrial quality control systems. ROS are generated in a number of cellular compartments, but most of the intracellular ROS can be traced back to the mitochondria.10, 11 There are eight sites within the mitochondrion that are known to possess the ability to generate ROS, the major source being complex 1.12, 13 In aging, the mitochondrial free-radical theory suggests that the progressive alteration of mitochondria that occurs with the aging process results in the increased production of ROS that in turn causes further mitochondrial dysfunction and damage to the entire cell.14 According to this theory, excessive production of ROS stimulates cytosolic signaling molecules that mediate the intrinsic mitochondrial apoptotic pathway.15
mtDNA mutation
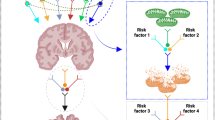
Mitochondria contain of the order of 1400 different proteins, but a vast number of these are encoded by the nuclear genome. Of the mitochondrial respiratory chain complex proteins, 13 that are required for adenosine triphosphate synthesis are encoded by the mitochondrial genome.16, 17 Recently, Kukat et al.18 reported structural organization of a single mtDNA molecule bound by the TFAM mitochondrial transcription factor using stimulated emission depletion microscopy. Mitochondrial DNA is known to have a higher mutation rate than nuclear DNA; this is because of multiple factors, including absence of protective histone molecules and the proximity of the mitochondrial genome to the inner mitochondrial membrane where ROS are routinely generated. However, mitochondria contain many antioxidant and DNA repair enzymes to prevent DNA mutation; these include OGG1 and MUTYH.19 Furthermore, the mitochondrion has its own mechanisms for regulating gene expression that are distinct from those of the nucleus. The mitochondrial genome is initially transcribed as a whole, before a large body of post-transcriptional machinery acts to generate individual gene products and to modulate their expression.20 Mitochondrial DNA quality control is important for communication with the nucleus. ROS-mediated gene expression that occurs upon oxidative phosphorylation dysfunction may result in a mitochondrial retrograde signaling pathway that can stimulate an adaptive nuclear response to mtDNA impairment. Mitochondrial genetic alterations induce a complex nuclear response that affects the expression of ⩾40 nuclear genes;21, 22 analysis suggested that, following treatment with various respiratory chain inhibitors or after mtDNA deletion, the disruption of mammalian mitochondrial function elicited a signaling response that involved calcineurin-dependent activation of nuclear factor-κB.23 Conversely, mtDNA dysfunction can be induced by many signaling molecules that are regulated by nuclear genes, and by factors related to mitochondrial metabolism. Many studies have shown that peroxisome proliferator-activated receptor-γ coactivator 1 and SIRT1 are involved in the regulation of mitochondrial function by activating the expression of nuclear genes such as TFAM24, 25 (Figure 1). Recently, Gomes et al. reported that nuclear nicotinamide adenine dinucleotide regulates mitochondrial transcription via a peroxisome proliferator-activated receptor-γ coactivator 1α/β-independent pathway.26 Furthermore, the tumor suppressor protein, p53, can regulate nuclear–mitochondrial communication via the mitochondrial disulfide relay system27 (Figure 1). Thus, mtDNA mutation is closely associated with nuclear signaling pathways and influences the process of aging.

Schematic model of the communication between mitochondria and the nucleus. Signaling between mitochondria and the nucleus is tightly controlled under cellular homeostasis. However, excessive reactive oxygen species (ROS) production induces translocation of the p53 protein to the mitochondria and suppression of peroxisome proliferator-activated receptor-γ coactivator 1; inhibition of the mitochondrial electron transport chain (ETC) by oxidative stress results in alteration of the nuclear genome. mtDNA, mitochondrial DNA; OXPHOS, oxidative phosphorylation.
Relationship between ROS and mtDNA dysfunction
Fusion and fission dysfunction
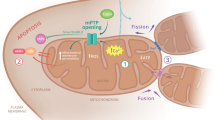
Mitochondria are especially dynamic organelles that are motile and that divide and fuse. These mitochondrial dynamics are critical for mitochondrial homeostasis and the maintenance of mitochondrial function. Whereas mitochondrial fusion allows mitochondria to combine and interact with each other, the opposite process, mitochondrial fission, facilitates mitochondrial rearrangement, remodeling and proliferation.28 Fusion and fission allow the incorporation of mtDNA and metabolites, the redistribution of mitochondria and cellular homeostasis in order to respond to energy demand (Figure 2). In yeast, fusion-deficient mutants fail to retain their mitochondrial genome and show defects in respiration.29, 30 Furthermore, mitochondrial dynamics are directly correlated with apoptosis. Some studies have demonstrated that mitochondrial fragmentation via the dynamin-related protein 1 (DRP1)-dependent pathway results in apoptosis in many organisms. Furthermore, the observation that the pro-apoptotic protein, BAX, interacts with DRP1 and mitofusins provides strong evidence for crosstalk between the mitochondrial dynamics machinery and apoptosis.31, 32 Many studies have demonstrated that excessive production of free radicals, including ROS and reactive nitrogen species, promotes neuronal cell damage in neurodegenerative disorders. Lipton and colleagues33 reported that S-nitrosylation of DRP1 at Cys644 contributed to amyloid-β (Aβ)-induced mitochondrial dynamics dysfunction, accelerating neuronal loss. Furthermore, Barsoum et al.34 reported that mitochondria undergo excessive fission induced by nitric oxide in cortical primary neurons. Recently, Byun et al.35 also demonstrated that ROS-mediated Sp1-Crif1 pathway is one of crucial mechanisms underlying mitochondrial dysfunction in AD. This evidence suggests that disruption of mitochondrial dynamics is an early event in ROS- or reactive nitrogen species-induced cell death.

Schematic model of the mitochondrial response to oxidative stress. Oxidative stress, including reactive oxygen species (ROS) and reactive nitrogen species, induces mitochondrial DNA (mtDNA) mutation and cellular dyshomeostasis. Oxidative stress and mtDNA mutation can lead to: (1) dysfunction of mitochondrial fusion and/or fission dynamics, (2) recruitment of the NLRP3 inflammasome and (3) mitochondria-associated endoplasmic reticulum membrane (MAM) alteration.
Inflammasomes
Inflammasomes are multiprotein complexes that form upon exposure to pathogenic microbes and host danger signals in order to activate caspase-1, leading to the maturation of interleukin-1β and interleukin-18. Inflammasome assembly serves as an activation platform for the proinflammatory caspases, caspase-1 and caspase-11.36 Inflammasome complexes contain NLRP1, NLRP3 and NLRC4, which are members of the NOD-like receptor family, together with the apoptosis-associated speck-like protein containing CARD (ASC) adaptor protein, all essential components of the inflammasome. NLRP3 is the crucial component of the inflammasome complex and it oligomerizes with the ASC adaptor protein and procaspase-1.37 ROS generation has been proposed to be one of the general features of NLRP3 activation. The induction of ROS is thought to lead to the generation of a possible ligand of NLRP3 or to directly affect NLRP3 or associated proteins. The assumption that mitochondrial ROS induces NLRP3 activation is based on the observation that most NLRP3 activators also cause ROS generation in immune cells such as macrophages and monocytes.38 Recent reports suggest a feasible role for mitochondrial recruitment of NLRP3 in inflammasome function, but this remains questionable. NLRP3 stimuli induced a translocation of NLRP3 from the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM), where it forms a functional inflammasome with caspase-1 and ASC.39 Some studies suggest mitochondria as the cellular source of the ROS and oxidized DNA that are required for NLRP3 activation (Figure 2). If interaction with mitochondria is important for NLRP3 function, how recruitment from the cytosol would be mediated is unclear; Subramanian et al.40 demonstrated that the mitochondrial antiviral signaling adaptor protein, MAVS, is needed, and that it acts to stimulate recruitment of NLRP3 to the mitochondria. They also suggest that factors other than MAVS may mediate mitochondrial recruitment of NLRP3 in response to inflammasome activators;40 for example, NLRP3 can be activated by a variety of stimuli, such as cholesterol, amyloid deposits, hydroxyapatite crystals, silica and fatty acids. The islet amyloid polypeptide gene product, IAPP, is a potential key activator of the NLRP3 inflammasome, and amyloid particles in general can activate the NLRP3 inflammasome. In addition, Wen et al.41 suggest that microglia-specific activation of the NLRP3 inflammasome is involved in AD pathogenesis. They showed that Aβ activates the NLRP3 inflammasome in microglia. Many studies have shown that diabetes and neurodegenerative disease share a pathogenic mechanism, and therefore it is possible that NLRP3 inflammasomes are key mediators of neuroinflammation.
The MAM
ROS are considered as key regulators of mitochondrial and ER function, and as activators of the unfolded protein response, in a variety of neurodegenerative diseases. How ROS regulate ER–mitochondria crosstalk during ROS-mediated cellular dysfunction has not been fully examined. The MAM establishes close contacts with the mitochondrion, supports translocation of lipids from the ER to the mitochondrion and regulates calcium homeostasis between these two organelles.42, 43 Recently, Hamasaki et al.44 demonstrated that autophagosomes form at the MAM, suggesting that the ER–mitochondria tether is essential in autophagosome formation and maintains physiological homeostasis. However, thus far, only a minority of MAM-regulating proteins (PACS2, HSPA9, σ1-receptor and mitofusin-2) are known. Verfaillie et al.45 reported an unexpected role for the ER stress sensor, RNA-dependent protein kinase R-like ER kinase, as a crucial component of the MAM; it can serve as a structural contact site at the ER–mitochondrion interface, coordinating crosstalk between these two organelles in ROS-induced cell death. Furthermore, Iwasawa et al.46 demonstrated that the FIS1 protein transfers an apoptotic signal from mitochondria to the ER by combining with the ER outer membrane protein, B-cell receptor-associated protein 31 (BAP31), and stimulating its cleavage to generate the p20BAP31 peptide fragment, the pro-apoptotic form of the BAP1 protein. They showed that the association between FIS1 and BAP31 is capable of triggering the apoptotic pathway (Figure 2). Continuous ER stress or the excessive generation of ROS induces the release of calcium from the ER at the MAM, causing cellular dyshomeostasis. These ROS-dependent MAM alterations can induce diverse effects in neurodegenerative disease, including AD. Area-Gomez et al.47 reported upregulated MAM function at the ER–mitochondrial interface, and increased crosstalk between these two organelles in AD; they proposed that MAM functions and ER–mitochondrial association can be upregulated by mutations in presenilin-1, presenilin-2 and the amyloid precursor protein, implying that presenilins are negative regulators of these phenomena. Hedskog et al.48 also reported increased expression of MAM-regulating proteins in AD models, although one of the MAM-associated proteins, the σ1-receptor, was significantly decreased in some AD mouse models, indicating diverse roles of MAM-associated proteins in neurodegenerative disease, and needing further examination.
Effects of mtDNA mutations on neurodegenerative diseases
Alzheimer’s disease
Mitochondrial accumulation of Aβ is one of the key mechanisms to cause mitochondrial dysfunction and lead to pathological process in AD.49 Mitochondrial genomic dysfunction is also reported in AD pathology. Damaged DNA lesions by oxidative stress are much higher in mtDNA of AD post-mortem tissues. Furthermore, degraded mtDNA and related proteins were found in the mitochondria of AD brains,50 and base excision repair (BER) pathway is defective in AD post-mortem brain whole tissue lysates.51 Krishnan et al.52 reported a higher percentage of cytochrome c oxidase-deficient neurons, which shows a higher level of mtDNA mutations, in the AD brains compared with age-matched controls. Swedlow et al. also found the mtDNA-depleted cells absence functional electron transport chains (ETCs) because they cannot produce mtDNA-encoded ETC components. In the AD cybrid cell lines, lowering cytochrome c oxidase activity, dysregulation of calcium homeostasis, increased ROS generation, reduction of mitochondrial membrane potentials, elevation of apoptotic pathways and increased Aβ42 production were reported.8 Their findings reveal that mtDNA mutation has crucial role for the AD-related mitochondrial dysfunction.
Parkinson’s disease
Many evidences report that mitochondrial dysfunction plays a crucial role in the pathogenesis of PD, relating that rotenone (complex 1 inhibitor) of the ETC complex can induce parkinsonism.53 Mitochondrial alteration because of oxidative damage, mtDNA mutations and mitochondrial dynamics dysfunction lead to degeneration of dopaminergic neurons.54 In addition, ETC deficiency in dopaminergic neurons leads to excessive mitochondrial fragmentation and an impaired supply of fresh mitochondria to dopaminergic nerve terminals in striatum.55 In addition, many studies suggest that PD-associated mitochondrial dysfunction is related to PTEN-induced putative kinase 1 (PINK1). Increasing excessive mitochondrial fission inhibits maintenance of oxidative phosphorylation (OXPHOS) machinery and loss of PINK1 results in a defective assembly of the OXPHOS complexes, leading to impaired mitochondrial OXPHOS.56, 57 In line with these observations, Kraytsberg et al.58 reported a high level of mtDNA deletion that is associated with ETC deficiency in substantia nigra neurons from PD patients. Pickrell et al.59 also demonstrated striatal dysfunctions in PD-mito-Pst1 mouse that expresses a mitochondrial restriction enzyme to damage mtDNA. These data suggest that mtDNA mutation can contribute to progression of PD.
Amyotrophic lateral sclerosis
The link between mitochondrial alteration and the progression of amyotrophic lateral sclerosis (ALS) is not fully understood yet, but some evidences imply that mutant SOD1 is the cause of mitochondrial dysfunction in ALS. Mutant SOD1 can inhibit VDAC1 (voltage-dependent anion-selective channel protein 1) directly, leading to reduction of energy production in mitochondria.60, 61 In the ALS but not control brains, levels of mtDNA were an average of more than 30-fold higher in the motor cortex, and elevated oxidative damage to mtDNA was detected in spinal motorneurons of the transgenic ALS mice.62, 63 Kikuchi et al.64 demonstrated that oxidative stress affects to the mitochondria and DNA repair mechanism are altered in ALS. Murakami et al.60 also reported that mitochondrial BER enzymes, ogg1 and pol-γ, are downregulated in mutant SOD1 transgenic mice. These data support a pathophysiologic role of mtDNA mutations in ALS.
Huntington’s disease
Mitochondrial dysfunction plays an important role in Huntington’s disease (HD) pathogenesis, as mitochondrial metabolic deficits and decreased activity of mitochondrial key enzymes have been found in HD patients. Furthermore, mutant huntingtin (Htt) or its polyQ-containing fragments can lead to mitochondrial dysfunction. Many other studies also suggested that mtDNA is a major target of mutant Htt-associated oxidative stress and may lead to mitochondrial alteration and that BER enzyme APE1 is one of the crucial target in the maintenance of mitochondrial activity in HD.65, 66 Siddiqui et al.67 demonstrated that mutant Htt-expressing cells exhibit higher mtDNA damage and reduced respiratory function than control cells. Furthermore, knockdown of APE1 results in mitochondrial dysfunction in mutant Htt-expressing cells. Wang et al.68 also suggested that HD cells, which have excessive mitochondrial Ca2+ levels, show higher level of mtDNA damage because of ROS generation. These studies provide the evidence for a link between mtDNA mutations and HD.
Defense mechanisms and therapeutic targets
Mitochondrial unfolded protein response and mitophagy
Mitochondrial quality control mechanisms are essential in order to maintain cellular homeostasis. In aging, nuclear and mitochondrial genomes are mutated, and damage occurs to intracellular organelles; mitochondrial quality control mechanisms can determine the consequences of this in terms of cell fate. Several quality control mechanisms remove damaged proteins or the entire organelle in order to sustain a functional mitochondrion. One of the major mechanisms for protein degradation within mitochondria is the ubiquitin–proteasome system.69 Excessive ROS production induces cellular dyshomeostasis, and mitochondria are vulnerable to the accumulation of damage. The mitochondrial unfolded protein response mitigates a large body of unfolded proteins in the mitochondria by triggering the induction of mitochondrial chaperone proteins, such as the mitochondrial chaperonin heat shock protein 60 (HSP60) and the HSPA9 protein. The triggering factor that activates the mitochondrial unfolded protein response is still questionable; it is important to preserve a balance between the expression levels of multiple proteins in the mitochondria, including those of the respiratory chain complex.70
Recent studies suggest that crosstalk between the mitochondria and autophagy has links to neurodegenerative disease. Autophagy has crucial roles in the adaptation to cellular stress, and as a quality control mechanism. Defects in autophagy are especially associated with neurodegenerative disorders that are in turn related to mitochondrial dysfunction.71 Nakahira et al.72 showed that autophagy defects render mitochondria dysfunctional, with consequent induction of the FGF21 protein as a mitokine. Recently, mounting evidence has implicated mitophagy and mitochondria-specific autophagic degradation as quality control mechanisms.73 Furthermore, Kurihara et al.74 reported that damaged mitochondria induce abnormal ROS generation that is related to mtDNA dysfunction in mitophagy-deficient cells, demonstrating that mitophagy is directly associated with the regulation of mitochondrial function.
Mitochondria-specific antioxidant therapy and gene therapy
Therapeutic approaches targeting mitochondria are attractive, because effects on a few general pathways can rescue malfunctions in a variety of ROS-mediated mitochondrial disorders. A possible approach is to target antioxidants to mitochondria via conjugation of a lipophilic cation, such as triphenylphosphonium. Lipophilic cations are capable of rapidly entering mitochondria in vivo because of the high mitochondrial membrane potential.75 Furthermore, this mitochondrial targeting approach can be used to enhance the therapeutic potential of an antioxidant in comparison with unconjugated antioxidants that exert effects elsewhere in the cell. In general, antioxidants have failed in clinical trials, because they do not have specificity for the mitochondrion, the organelle that is most vulnerable to the pathogenic effects of ROS.76 Mitochondria-targeted antioxidants are capable of a 100-fold accumulation within mitochondria that contributes to more effective protection against cellular damage; for example, MitoQ can be selectively targeted to mitochondria, and directly protects them from ROS.77 Another mitochondria-specific antioxidant, SS31, which has an innate antioxidant function, has been demonstrated to be effective in a variety of animal models, and is now being tested in clinical trials. As reported by McManus et al.,78 MitoQ mitigated cognitive decline, as well as oxidative damage, astrogliosis, Aβ accumulation and neuronal loss, in an AD mice model; MitoQ was also able to mitigate oxidative stress and prevent cell death in other disease models. MitoQ and SS31, which are taken up by the inner mitochondrial membrane, significantly improved neurite outgrowth and synaptic function in neurons affected by AD.79, 80 These findings indicate that MitoQ and SS31 reduce Aβ-mediated mitochondrial dysfunction (Figure 3).

Mitochondrial defense strategies. Mitochondria have internal defense mechanisms, such as the unfolded protein response (UPR) and mitophagy. However, excessive reactive oxygen species (ROS) generation and mitochondrial DNA (mtDNA) mutation result in the inhibition of these systems. Mitochondria-specific antioxidants and mitochondria-targeted genomic modulation can reduce oxidative stress-induced mtDNA mutation and mitochondrial dysfunction.
Recently, several approaches have been applied to mitigate mutant mtDNA-mediated cellular damage. BER enzymes, which are all nuclear encoded and imported into the mitochondria, are primarily responsible for removing oxidized DNA bases, including 8-oxo-2′-deoxyguanosine, from mtDNA. Tonin et al.81 developed specially constructed RNA molecules containing key structural domains for mitochondrial import and corresponding to the mutated region of mtDNA, and demonstrated that these were able to specifically anneal to mutated mitochondrial genomes; these recombinant RNA molecules induced a decrease in the proportion of mtDNA molecules bearing a pathogenic mutation. Gammage et al.82 designed and engineered mitochondria-specific heterodimeric zinc-finger nucleases for site-specific elimination of pathogenic human mtDNA. Kim et al.83 demonstrated that mitochondria-targeted human OGG1, a key enzyme in BER, is necessary for preventing mtDNA dysfunction. Bacman et al.84 engineered transcription activator-like effector nucleases to selectively target mitochondria and cleave multiple pathogenic mtDNA mutations (Figure 3). These various gene therapy approaches, which can potentially rescue ROS-induced mtDNA dysfunction in mitochondrial disease, might also be useful in neurodegenerative disease.
Conclusion
Thus far, mitochondrial genome studies have not investigated the link between mtDNA alteration and mitochondrial abnormality, although loss of mitochondrial genomic integrity has been implicated in many neurodegenerative diseases. As mitochondria are vulnerable to oxidative damage, it is possible that a feedback loop, involving oxidative damage and mitochondrial dysfunction, contributes to the initiation and/or amplification of ROS production that is a key player in the pathogenesis of a variety of diseases. As mitochondria communicate with other organelles, such as the nucleus and ER, mutations in mtDNA that result in stable perturbations of mammalian mitochondrial function can also elicit a coordinated alteration in nuclear gene expression.85 In addition, mitochondrial dysfunction, including excessive activation of the fission and fusion machinery, results in MAM alteration and recruitment of the NLRP3 inflammasome. Mitochondria are crucial for the maintenance of cellular homeostasis, and abnormal oxidative stress impairs mitochondrial defense systems. In line with this observation, it is important to prevent mtDNA dysfunction as a possible way to delay the progress of neurodegenerative disease.
References
Swerdlow RH, Burns JM, Khan SM . The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 2013; 1842: 1219–1231.
Calkins MJ, Reddy PH . Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from Alzheimer’s disease mice: implications for impaired mitochondrial biogenesis and synaptic damage. Biochim Biophys Acta 2011; 1812: 1182–1189.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 2001; 21: 3017–3023.
Gredilla R, Weissman L, Yang J-L, Bohr VA, Stevnsner T . Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer’s disease. Neurobiol Aging 2012; 33: 694–707.
Miquel J . An update on the mitochondrial-DNA mutation hypothesis of cell aging. Mutat Res 1992; 275: 209–216.
Readnower RD, Sauerbeck AD, Sullivan PG . Mitochondria, amyloid β, and Alzheimer’s disease. Int J Alzheimers Dis 2011; 2011: 104545.
Lezza AM, Boffoli D, Scacco S, Cantatore P, Gadaleta MN . Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem Biophys Res Commun 1994; 205: 772–779.
Yan MH, Wang X, Zhu X . Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 2013; 62: 90–101.
Lin MT, Beal MF . Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443: 787–795.
Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E . Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 2012; 322: 254–262.
Lagouge M, Larsson N-G . The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med 2013; 273: 529–543.
Oxygen R, Nijtmans LGJ, Dieteren CEJ, Roestenberg P, Valsecchi F, Smeitink JAM et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal 2010; 12: 1431–1470.
Brand MD . The sites and topology of mitochondrial superoxide production. Exp Gerontol 2010; 45: 466–472.
López-Otín C, Blasco Ma, Partridge L, Serrano M, Kroemer G . The hallmarks of aging. Cell 2013; 153: 1194–1217.
Circu ML, Aw TY . Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010; 48: 749–762.
Spelbrink JN . Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life 2010; 62: 19–32.
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J et al. Sequence and organization of the human mitochondrial genome. Nature 1981; 290: 457–465.
Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N-G, Jakobs S . Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA 2011; 108: 13534–13539.
Reeve AK, Krishnan KJ, Turnbull D . Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann NY Acad Sci 2008; 1147: 21–29.
Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood A-MJ et al. The human mitochondrial transcriptome. Cell 2011; 146: 645–658.
Epstein CB, Waddle JA, Hale W, Davé V, Thornton J, Macatee TL et al. Genome-wide responses to mitochondrial dysfunction. Mol Biol Cell 2001; 12: 297–308.
Yun J, Finkel T . Mitohormesis. Cell Metab 2014; 19: 757–766.
Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG . Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol 2003; 161: 507–519.
Finley LWS, Haigis MC . The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev 2009; 8: 173–188.
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–273.
Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013; 155: 1624–1638.
Zhuang J, Wang P-Y, Huang X, Chen X, Kang J-G, Hwang PM . Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci USA 2013; 2: 2–7.
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E . Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 2008; 9: 505–518.
Westermann B . Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010; 11: 872–884.
Merz S, Westermann B . Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol 2009; 10: R95.
Suen D, Norris K, Youle R . Mitochondrial dynamics and apoptosis. Genes Dev 2008;: 1577–1590.
Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA 2007; 104: 11649–11654.
Nakamura T, Lipton SA . Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ 2011; 18: 1478–1486.
Barsoum MJ, Yuan H, Gerencser Aa, Liot G, Kushnareva Y, Gräber S et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006; 25: 3900–3911.
Byun J, Son SM, Cha M-Y, Shong M, Hwang YJ, Kim Y et al. CR6-interacting factor 1 is a key regulator in Aβ-induced mitochondrial disruption and pathogenesis of Alzheimer’s disease. Cell Death Differ, (e-pub ahead of print 7 November 2014; doi:10.1038/cdd.2014.184.
Franchi L, Muñoz-Planillo R, Núñez G . Sensing and reacting to microbes through the inflammasomes. Nat Immunol 2012; 13: 325–332.
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G . The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009; 10: 241–247.
Lawlor KE, Vince JE . Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta 2013; 1840: 1433–1440.
Brown GC, Bal-Price A . Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 2003; 27: 325–355.
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN . The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013; 153: 348–361.
Wen H, Ting JP-Y, O’Neill LAJ . A role for the NLRP3 inflammasome in metabolic diseases—did Warburg miss inflammation? Nat Immunol 2012; 13: 352–357.
Raturi A, Simmen T . Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 2013; 1833: 213–224.
Simmen T, Lynes EM, Gesson K, Thomas G . Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta 2010; 1798: 1465–1473.
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495: 389–393.
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere J-P et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19: 1880–1891.
Iwasawa R, Mahul-Mellier A-L, Datler C, Pazarentzos E, Grimm S . Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 2011; 30: 556–568.
Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJC, Madra M et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 2012; 31: 4106–4123.
Hedskog L, Moreira C, Filadi R, Rönnbäck A, Hertwig L, Wiehager B . Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci USA 2013; 110: 7916–7921.
Cha M-Y, Han S-H, Son SM, Hong H-S, Choi Y-J, Byun J et al. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One 2012; 7: e34929.
Reddy PH . Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol 2009; 218: 286–292.
Canugovi C, Shamanna RA, Croteau DL, Bohr VA . Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol Aging 2014; 35: 1–8.
Krishnan KJ, Ratnaike TE, De Gruyter HLM, Jaros E, Turnbull DM . Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol Aging 2012; 33: 2210–2214.
Winklhofer KF, Haass C . Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta 2010; 1802: 29–44.
Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG . Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta 2013; 1842: 1282–1294.
Sterky FH, Lee S, Wibom R, Olson L, Larsson N-G . Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci USA 2011; 108: 12937–12942.
Liu W, Acín-Peréz R, Geghman KD, Manfredi G, Lu B, Li C . Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci USA 2011; 108: 12920–12924.
Tufi R, Gandhi S, de Castro IP, Lehmann S, Angelova PR, Dinsdale D et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat Cell Biol 2014; 16: 157–166.
Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K . Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 2006; 38: 518–520.
Pickrell AM, Pinto M, Hida A, Moraes CT . Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 2011; 31: 17649–17658.
Murakami T, Nagai M, Miyazaki K, Morimoto N, Ohta Y, Kurata T et al. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res 2007; 1150: 182–189.
Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010; 67: 575–587.
Dhaliwal GK, Grewal RP . Mitochondrial DNA deletion mutation levels are elevated in ALS brains. NeuroReport 2000; 11: 2507–2509.
Warita H, Hayashi T, Murakami T, Manabe Y, Abe K . Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res 2001; 89: 147–152.
Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T . Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol 2002; 103: 408–414.
Ayala-Peña S . Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic Biol Med 2013; 62: 102–110.
Oliveira JMa, Lightowlers RN . Could successful (mitochondrial) networking help prevent Huntington’s disease? EMBO Mol Med 2010; 2: 487–489.
Siddiqui A, Rivera-Sánchez S, Castro Mdel R, Acevedo-Torres K, Rane A, Torres-Ramos CA et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic Biol Med 2012; 53: 1478–1488.
Wang J-Q, Chen Q, Wang X, Wang Q-C, Wang Y, Cheng H-P et al. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J Biol Chem 2013; 288: 3070–3084.
Segref A, Kevei E, Pokrzywa W, Schmeisser K, Mansfeld J, Livnat-Levanon N et al. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab 2014; 19: 642–652.
Andreux Pa, Houtkooper RH, Auwerx J . Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 2013; 12: 465–483.
Okamoto K . Mitochondria breathe for autophagy. EMBO J 2011; 30: 2095–2096.
Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Green DR, Galluzzi L, Kroemer G . Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333: 1109–1112.
Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T et al. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287: 3265–3272.
Smith RaJ, Hartley RC, Cochemé HM, Murphy MP . Mitochondrial pharmacology. Trends Pharmacol Sci 2012; 33: 341–352.
Dai D-F, Rabinovitch PS, Ungvari Z . Mitochondria and cardiovascular aging. Circ Res 2012; 110: 1109–1124.
Murphy MP, Smith RAJ . Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 2007; 47: 629–656.
McManus MJ, Murphy MP, Franklin JL . The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci 2011; 31: 15703–15715.
Rodriguez-Cuenca S, Cochemé HM, Logan A, Abakumova I, Prime TA, Rose C et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med 2010; 48: 161–172.
Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP et al. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis 2010; 20 (Suppl 2): S609–S631.
Tonin Y, Heckel A-M, Vysokikh M, Dovydenko I, Meschaninova M, Rotig A et al. Modelling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mtDNA. J Biol Chem 2014; 289: 13323–13334.
Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M . Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med 2014; 6: 1509–1639.
Kim S-J, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT et al. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem 2014; 289: 6165–6176.
Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT . Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med 2013; 19: 1111–1113.
Heddi A, Stepien G, Benke PJ, Wallace DC . Coordinate induction of energy gene expression in tissues of mitochondrial disease patients. J Biol Chem 1999; 274: 22968–22976.
Acknowledgements
This work was supported by grant from NRF 2012R1A2A1A01002881 and by Protein Metabolism Medical Research Center through Seoul National University Nobel Laureates Invitation program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Cha, MY., Kim, D. & Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med 47, e150 (2015). https://doi.org/10.1038/emm.2014.122
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.122
This article is cited by
-
Mitochondrial DNA quantification correlates with the developmental potential of human euploid blastocysts but not with that of mosaic blastocysts
BMC Pregnancy and Childbirth (2023)
-
Adiponectin deficiency accelerates brain aging via mitochondria-associated neuroinflammation
Immunity & Ageing (2023)
-
The contributions of mitochondrial and nuclear mitochondrial genetic variation to neuroticism
Nature Communications (2023)
-
Zbp1 gene: a modulator of multiple aging hallmarks as potential therapeutic target for age-related diseases
Biogerontology (2023)
-
Mitochondrial Dysfunction as a Signaling Target for Therapeutic Intervention in Major Neurodegenerative Disease
Neurotoxicity Research (2023)
