
Abstract
The maintenance of whole-body glucose homeostasis is critical for survival, and is controlled by the coordination of multiple organs and endocrine systems. Pancreatic islet β cells secrete insulin in response to nutrient stimuli, and insulin then travels through the circulation promoting glucose uptake into insulin-responsive tissues such as liver, skeletal muscle and adipose. Many of the genes identified in human genome-wide association studies of diabetic individuals are directly associated with β cell survival and function, giving credence to the idea that β-cell dysfunction is central to the development of type 2 diabetes. As such, investigations into the mechanisms by which β cells sense glucose and secrete insulin in a regulated manner are a major focus of current diabetes research. In particular, recent discoveries of the detailed role and requirements for reorganization/remodeling of filamentous actin (F-actin) in the regulation of insulin release from the β cell have appeared at the forefront of islet function research, having lapsed in prior years due to technical limitations. Recent advances in live-cell imaging and specialized reagents have revealed localized F-actin remodeling to be a requisite for the normal biphasic pattern of nutrient-stimulated insulin secretion. This review will provide an historical look at the emergent focus on the role of the actin cytoskeleton and its regulation of insulin secretion, leading up to the cutting-edge research in progress in the field today.
Similar content being viewed by others


Introduction
Biphasic insulin secretion from pancreatic islet β cells
In response to increased blood glucose levels, pancreatic islet β cells secrete insulin in a biphasic manner (Figure 1). Glucose enters the β cell and is rapidly metabolized to yield a net increase in the ATP/ADP ratio. ATP-sensitive potassium (KATP) channels close and cause membrane depolarization, which opens voltage-dependent calcium channels.1 Subsequent calcium influx leads to insulin granule docking and fusion with the plasma membrane in a process referred to as ‘insulin granule exocytosis’.2, 3, 4 Insulin granule docking and fusion events are mediated by a group of proteins termed soluble N-ethylmaleimide sensitive factor attachment receptor (SNARE) proteins. This stimulus-secretion coupling pathway results in a rapid robust spike of insulin secretion called first phase, derived from insulin granules that are both pre-docked within 100–200 nm of the plasma membrane as well as so-called ‘restless newcomer’ granules; this pool of release-competent granules is referred to as the ‘readily releasable pool’.5, 6 After the first-phase peak, the insulin release rate drops to two- to fivefold above basal secretion and is sustained at this lower rate of insulin release, called ‘second phase,’ which persists until euglycemia is restored.7, 8

Biphasic glucose-stimulated insulin secretion from perfused mouse islets. In response to a square-wave increase in glucose concentration, islet β cells dock/fuse ∼50–100 granules in the first phase of secretion. First phase is temporally defined, complete within ∼10 min of glucose stimulation, thought to be accounted for in large part by the readily releasable pool of granules. First phase is immediately followed by a second phase, with second phase being lower in amplitude but persistent in the presence of glucose stimulation over hours of time, and thus not temporally limited. Second phase is presumed to require storage/reserve pools of granules that can be recruited to the plasma membrane for secretion.
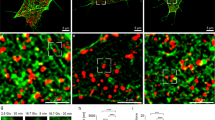
Second-phase insulin secretion requires the recruitment of granules from intracellular storage pools to the plasma membrane and involves reorganization of the filamentous actin (F-actin) cytoskeletal network (Figure 2a).9, 10, 11 This reorganization takes place in the cortical F-actin web (Figure 2b). Stress fiber-localized actin and focal adhesion actin-remodeling events, which occur at cell surface and cell–cell junctions, also contribute to insulin secretion.12, 13, 14 This review will focus largely on cortical F-actin remodeling, as it is cortical F-actin that oversees insulin granule access to the plasma membrane, but the importance of focal adhesions will be discussed where relevant. Pre-clinical and frank type 2 diabetes are associated with losses in both first and second phases of insulin secretion.15, 16 Of the two phases, there is far less known about the mechanistic regulation of second-phase insulin secretion. Key proteins shown by perfusion to be required for both phases of insulin secretion include, but are not necessarily limited to, the SNARE protein Syntaxin 4, the SNARE-related proteins tomosyn-2 and Doc2b, Rab27a, Rab3a, granuphilin and RalA.17, 18, 19, 20, 21, 22 The SNARE protein Syntaxin 1 is only required for first phase.23 Currently identified regulators specific to second-phase insulin secretion include Cdc42, p21-activated kinase (PAK1), RhoGDI, oleate/GPR40/PKD1 and Munc18c.24, 25, 26, 27, 28 Notably, many proteins required for biphasic secretion have connections to actin cytoskeleton remodeling.

Cortical filamentous actin (F-actin) remodeling in β cells regulates insulin granule exocytosis. (a) Schematic of a pancreatic β cell depicts how readily releasable pool (RRP) granules are docked at the membrane and reserve pool granules are in a more intracellular storage pool. Cortical F-actin can regulate access of reserve pool granules to the readily releasable pool. (b) Microscopic analysis of β cell-actin remodeling is often performed on cells attached to coverslips (a very different environment from the three-dimensional islet architecture) where it is important to note that focal adhesions/stress fibers form at the cell attachment interface. Conversely, cortical F-actin is visualized at the cell perimeter when focused at the mid plane of the cell. This distinction is particularly important in differentiating cortical F-actin remodeling from other types of actin remodeling.
Filamentous actin as a regulator of insulin granule exocytosis
F-actin was first recognized as a regulator of exocytosis ∼40 years ago.29, 30, 31, 32, 33 Over this time, use of F-actin-disrupting drugs like cytochalasins, latrunculins, clostridium C2 toxins and F-actin-polymerizing drugs like phallotoxins and jasplakinolide, have proven useful in implicating roles for F-actin in secretory/exocytotic processes across multiple cell types.29, 33, 34 These cell types include pancreatic β cells and acinar cells,35 enteroendocrine L cells,36 chromaffin and PC12 cells,37, 38 melanotrophs and lactotropes,39, 40 neutrophils,41, 42, 43 platelets,44 endothelial cells,45 neurons,46 adipocytes47 and myocytes.48 In chromaffin cells, F-actin was shown to form a cortical ring beneath the plasma membrane, function as a barrier between vesicles and the cell surface, and to colocalize with cytoskeletal proteins such as myosin, caldesmon,30, 49, 50 gelsolin or scinderin 51 and fodrin.52 Landmark work in 1972 by Orci et al.29 showed electron micrographs of islet β cells depicting F-actin in the form of a cell web just beneath the plasma membrane, further demonstrating that depolymerization of F-actin with Clostridium botulinum C2 toxin, Cytochalasin B or E potentiated glucose-stimulated insulin secretion from pancreatic islets,33, 53, 54, 55 consistent with the ‘barrier’ model formulated in chromaffin cells. However, the role of F-actin as a simple barrier in the β cell was quite controversial, given additional data showing that actin depolymerization inhibited glucose-stimulated insulin secretion from the HIT (hamster) β cell line,33 as well as an increased fraction of F-actin in pancreatic β-cell homogenates prepared from rodent islets stimulated with glucose.56, 57 Technological advances made in the last decade enabling the visualization of F-actin remodeling and insulin exocytosis in β cells have re-invigorated this field of research, as has the outcomes of genome-wide association studies pointing to β cell functional failure as a primary feature of type 2 diabetes.
Positive and negative roles of F-actin in glucose-stimulated insulin exocytosis
To sustain insulin release, mature insulin granules located in the intracellular storage pools must be mobilized toward the plasma membrane. This process coincides with what is now referred to as glucose-induced remodeling of the actin cytoskeleton,56, 58 as it encompasses the simultaneous localized depolymerization and polymerization of F-actin across the cell in a concerted manner. The concept of remodeling has evolved from early studies wherein F-actin was first thought to function solely as a negative barrier to restrict insulin granule accumulation at the plasma membrane.29, 30, 33, 54 This was based upon a body of literature that relied upon pharmacological depolymerizing agents, usage of which results in more morphologically docked granules and may confer enhanced release competence to granules.10, 59 However, early evidence of positive effects of the cytoskeleton in stimulus-induced insulin secretion existed as well, raising controversy as to the true role and requirement for F-actin in insulin granule exocytosis.56, 57, 60, 61, 62 Upon the advent of live-cell and time-lapse imaging of subcellular-localized β cell F-actin changes using confocal microscopy, and in conjunction with genetic manipulations of proteins involved in F-actin changes rather than reliance upon pharmacological agents, these positive and negative roles of F-actin in insulin granule exocytosis were understood to be the cyclic nature of F-actin remodeling.
F-actin and the regulation of basal insulin secretion
Control of basal insulin secretion, the amount of insulin secreted under non-stimulatory or sub-threshold conditions, is a critical part of maintaining whole-body glucose homeostasis.63 Aberrantly increased insulin secretion under fasting conditions can cause hypoglycemia acutely, and chronically may contribute to the development of insulin resistance in peripheral tissues.63 The barrier role of F-actin is considered an integral part of maintaining low levels of insulin release under basal conditions, via restricting insulin granule access to the cell surface/plasma membrane docking and fusion machinery.
The actin cytoskeleton has been linked to roles in basal exocytosis through several factors: β-Pix,64 Caveolin-1,65 focal adhesion kinase (FAK),14 EphA–Ephrin–A signaling62 and Gelsolin.66 Depletion from β cells of either β-Pix (also called Cool-1) or Caveolin-1, which coordinately regulate Cdc42 activation, leads to dysregulated basal insulin secretion.64, 65 Cdc42 is a small Rho family guanine nucleotide triphosphates (GTPase) that is kept mostly inactive (>90%) in the β cell by the binding of Caveolin-1, the guanine nucleotide dissociation inhibitor (GDI) for Cdc42 localized to the insulin secretory granule. Cdc42 becomes activated in response to glucose stimulation by the dissociation of Caveolin-1 and the activation of the guanine nucleotide effector protein β-Pix (discussed in detail in the next subsection). A body of work suggests that maintenance of Cdc42 in its inactive state is important to maintain low levels of insulin secretion under basal (∼5 mM glucose) conditions. Pharmacological inhibition of FAK, localized to focal adhesions, caused an increase in basal insulin secretion, which was related to disrupted β cell F-actin structure due to discontinuous cell–cell contact.12, 14 EphA5–Ephrin–A5 signaling mediates a paracrine signaling pathway that regulates insulin secretion. Disruption of EphA5–Ephrin–A5 signaling in MIN6 β cells prevents normal glucose-induced Rac1 activation and F-actin remodeling.62 The binding of Gelsolin, a cortical F-actin severing and capping protein, to the docking and fusion target-SNARE protein, Syntaxin 4, required for biphasic insulin exocytosis, has been shown to be essential in the maintenance of minimal insulin release under basal conditions.66 While it is clear that signaling related to F-actin regulation has impacts on basal insulin secretion, further investigation of the dynamics of F-actin remodeling and how basal insulin secretion is controlled will be useful in understanding this linkage.
Signaling pathways leading to F-actin remodeling in the β cell
Small Rho family GTPases regulate F-actin remodeling and insulin secretion
Small GTPases, such as Cdc42 as mentioned above, have many roles in cell biology including, but not limited to cytoskeletal reorganization, membrane trafficking and cell growth.67 These GTPases are known as the Ras superfamily due to their sequence homology; well-studied families include Ras, Rho, Rab, Arf, Rap and Ran.68 Of particular importance to second-phase insulin release and F-actin remodeling is the Rho family, which contains protein members Cdc42, Rac and Rho. Small GTPases become activated when bound to GTP and remain active until the GTP is hydrolyzed to yield GDP and inorganic phosphate.67 The GDI-bound GTPase is restricted from the cell surface, and upon dissociation of the GDI from the GTPase enters the cycle of GTP binding and hydrolysis, whereby the guanine nucleotide effector proteins then promote the dissociation of GDP and binding of GTP.69 Active small GTPases bind to effector molecules to propagate their signal. As described in greater detail below (in the subsequent four sections), the activation of Cdc42 in β cells leads to subsequent activation of Rac1, through the activation/phosphorylation of the PAK1.25 Arf6,70 Rab27A,71, 72, 73, 74 Rab3a22 and Rap175 have emerged as additional potential positive effectors of insulin release. On the other hand, initial findings suggest against positive roles for RhoA, Ras or Ran in insulin secretion.76, 77
The Cdc42–PAK1–Rac1 signaling pathway
Early studies implicating Cdc42 and Rac1 cycling in F-actin remodeling coupled to glucose-stimulated insulin secretion involved expression of GTPase cycling mutants.34, 78 Subsequent studies using RNAi-mediated knockdown of each GTPase confirmed their roles to be in the second phase of insulin secretion, and further revealed their signaling itineraries in human and mouse β cells.25, 27 Most recently, β cell-specific Rac1 knockout mice have been characterized and the role of Rac1 in F-actin remodeling and second-phase insulin release confirmed.79 However, β cell-specific Cdc42 knockout mice have yet to be generated, and as Cdc42 was found to be required for formation of the pancreatic architecture,80 islets of classic Cdc42 knockout mice are unavailable for study.
In mouse insulinoma MIN6 and rat insulinoma INS 832/13 clonal β cell lines, Cdc42 is activated within 3 min of glucose stimulation and activation declines to basal levels within the following 2 min;25, 34, 64, 70 in human islets, this has been detected indirectly by the phosphorylation and activation of Cdc42’s downstream effector, PAK127, 81 (Figure 3). The activation–deactivation cycling of Cdc42 coincides with post-translational modifications such as carboxymethylation, prenylation and O-glycosylation.34, 82 The direct signaling factor responsible for activating Cdc42 remains at-large, although progress has been made in this regard. It has been deduced that Cdc42 activation requires glucose metabolism, as non-metabolizable glucose analogs or KCl-depolarization induced Ca2+ influx fail to induce Cdc42 activation.25 New data from our laboratory suggest this factor signals through a Src family kinase upstream of Cdc42 activation (unpublished results, SM Yoder and DC Thurmond), wherein activation follows with the glucose-stimulated association of Cdc42 with β-Pix within 2 min, coordinate with the timing of Cdc42–GDP dissociation from its GDI, Caveolin-1.64 Upon activation, Cdc42 triggers phosphorylation and activation of PAK1, ∼5 min from the start of glucose stimulation, followed by Rac1 activation after ∼10 min more (15 min from the start of glucose stimulation).25 Thus, the activation of Rac1 occurs during the second phase of insulin release, and both Cdc42 and Rac1 are required for this phase of secretion through activation of PAK1 in a sequential pathway.

Pathways impinging specifically on glucose-mediated F-actin remodeling in the β cell. The β cell responds to certain insulin secretagogues by remodeling its cortical filamentous actin (F-actin). Glucose metabolism can signal to Cdc42, at least partially though β-Pix, to activate p21-activated kinase (PAK1) and evoke Rac1 activation. Under basal conditions, Cdc42 and Rac1 are held inactive by guanine nucleotide dissociation inhibitor (GDI) proteins Caveolin-1 and RhoGDI. In addition, GLP-1 activation of GLP-1R can lead to increased cAMP and PKA activation which can activate SAD-A kinase and feed into the PAK1 pathway. Active PAK1 signals to multiple effectors, such as Rac1, to facilitate F-actin remodeling. PAK1 is important for Raf/MEK activation, leading to ERK activation. PAK1 (and possibly ERK as well) may then signal to myosin light-chain kinase (MLCK)-myosin IIA (MyoIIA) to further mediate F-actin remodeling. Focal adhesion kinase (FAK) participates in cross-talk with ERK, which may indirectly modulate cortical F-actin remodeling. The F-actin binding and severing protein Gelsolin complexes directly with the t-SNARE (soluble N-ethylmaleimide sensitive factor attachment receptor) protein Syntaxin 4, ultimately impacting SNARE-mediated insulin exocytosis. Syntaxin 4 in particular is essential for second-phase insulin release, complexing with the other t-SNARE protein synaptosomal-associated protein of 25 kDa (SNAP-25) (or SNAP-23) and the incoming granule vesicle-SNARE, VAMP2.
Under basal unstimulated conditions, activated Cdc42 is maintained at a low level, ∼7% of total Cdc42, via sequestration by GDIs;26, 34, 65 Rac1 is also kept in the inactive state.78 So far, two GDI proteins for Cdc42 in β cells have been identified: Caveolin-1 and RhoGDI.26, 65, 83 In the β cell, Cdc42 is in the cytosol, at the plasma membrane and on insulin secretory granules;64, 65 Rac1 is principally cytosolic.83 Caveolin-1, a membrane-bound protein important for caveolae formation as well as in signal transduction,84, 85, 86 was found to function in an unconventional manner as a Cdc42 GDI protein in islet β cells in 2006,65 binding to that quotient of Cdc42 localized to insulin granules; this new role for Caveolin-1 has since been confirmed in other cell types.87, 88 Conversely, RhoGDI binds to the cytosolic pool of Cdc42; RhoGDI also binds to Rac1 in the cytosol, and differential RhoGDI tyrosine and serine phosphorylation events account for the distinct spatial and temporal dissociation patterns of RhoGDI-Cdc42 versus RhoGDI-Rac1 in the β cell.26, 83 Depletion of RhoGDI from mouse islets selectively and robustly potentiates the second phase of insulin secretion, correlating with increased glucose-stimulated Cdc42 activation.26, 83 Whether it is the cytosolic pool of Cdc42 that is released from RhoGDI or the insulin granule pool of Cdc42 that is released from Caveolin-1 that ultimately activates PAK1 to then activate Rac1 remains to be discerned. Some evidence suggests that the cytosolic Cdc42 released from RhoGDI upon glucose stimulation transits to the insulin granule storage pool rather than to the plasma membrane (Z Wang and DC Thurmond, unpublished results). Cdc42 may be needed on the insulin granule to assist with their targeting to fusion sites at the plasma membrane for secretion, by virtue of its propensity to interact directly with the vesicle-SNARE protein VAMP2, forming heterotrimeric complexes with Syntaxins.89 While it initially strikes odd that Cdc42 activation occurs early during the first phase of insulin release but is only required selectively for second-phase glucose-stimulated insulin secretion, we speculate that the early activation of Cdc42 functions to initiate signaling cascades necessary to evoke F-actin remodeling and granule mobilization to the plasma membrane to support the second phase of insulin release. This is consistent with the multiple reports of F-actin remodeling visualization. Indeed, recent live-cell imaging demonstrates that PAK1 activation is essential for F-actin remodeling to occur in β cells.81 In this manner, the early activation of Cdc42 could facilitate insulin granule trafficking toward the plasma membrane, and/or enhance the release competence of granules that are already juxtaposed to the plasma membrane.
The role of PAK1 signaling in the β cell
PAK1 has recently evolved as a protein of interest in β cell function, as it is ∼80% reduced in the islets of type 2 diabetic individuals.27 Whether this deficit marks PAK1 as a point of genetic susceptibility to type 2 diabetes, or rather that PAK1 is affected as a consequence of metabolic aberrations associated with type 2 diabetes, remains to be determined. PAK1 is one of six members of the PAK family, with isoforms grouped into two main categories based upon sequence homology. Group I PAKs include PAK1, PAK2 and PAK3; group II includes PAK4, PAK5 and PAK6. The PAK (p21-activated kinase) family is named for the 21 kDa proteins that activate them, Rac1 and Cdc42. Both Cdc42 and Rac1 interact with the CRIB domain of PAK1, which in turn induces a conformational change in PAK1 to dissociate the kinase and regulatory domains to induce its activation.90 PAK1 can serve as the effector or upstream activator of Cdc42 and Rac1, with its role being seemingly cell-type dependent. For example, in β cells, PAK1 is activated downstream of Cdc42 but is the upstream activator of Rac125 (Figure 3). In non-β cells, Rac1 can be upstream of Cdc42,91 or Rac1 and Cdc42 can exert antagonistic actions.92 Recently PAK1 has been shown to be activated by SAD-A kinase (also known as BRSK2) in response to cAMP signaling in the β cell.93, 94 However, whether SAD-A kinase action on PAK1 is dependent on Cdc42 or not remains an open question, as does the true role of SAD-A kinase, as knockdown of this gene has been shown to both increase and decrease insulin secretion.93, 95
The recent availability of PAK1 knockout mice and of the PAK1 activation inhibitor IPA3 have helped to elucidate the role and requirement for PAK1 in second-phase secretion and F-actin remodeling in the islet β cell. Classic whole-body PAK1 knockout mice exhibit in vivo glucose intolerance,27, 93 and their islets show loss of second-phase insulin secretion ex vivo.27 Both human and mouse islets also exhibit glucose-stimulated ERK1/2 activation27, 81, 96, 97 and studies of islets from PAK1 knockout mice as well as PAK1-depleted clonal β cells revealed ERK1/2 activation to be dependent upon PAK1 activation and signaling (as will be discussed further in the next section). Alternatively, or in addition to signaling through ERK, PAK1 in other cell types has been demonstrated to utilize several other substrates for phosphorylation, including LIM kinase,98 Filamin A,99 myosin light-chain kinase (MLCK),100 RhoGDI101 and the p41-Arc subunit of the Arp2/3 complex.102 As such, it remains possible that the modification of one or more of these targets by PAK1 could contribute to F-actin remodeling and second-phase insulin secretion. As the MLCK substrate myosin IIA has recently been implicated in β cell-actin remodeling and insulin secretion,103 it is possible this pathway is linked through Cdc42-PAK1 signaling.
The role of Rac1 signaling in the β cell
Rac1 was first implicated in glucose-specific insulin secretion in 1997 in clonal β cells,76, 104 later confirmed to being refractory to KCl-stimulation in 2004.78 In 2007, Rac1 was placed downstream of Cdc42 activation, and in 2010, Kowluru et al.105 demonstrated that Raf-1 and ERK were required for glucose-induced Rac1 activation in INS 832/13 cells. Most recently, in 2012, characterizations of β cell-specific Rac1 knockout mice confirmed these results: the mice exhibited impairments in glucose tolerance in vivo, their islets showed impaired second-phase insulin release ex vivo with defective glucose-stimulated F-actin remodeling, and the islets showed no impairment to either first-phase glucose-stimulated or KCl-stimulated insulin release,79 confirming all prior clonal β cell studies. However, the question still remains about how and why Rac1 activation is detected after the initial visualization of F-actin remodeling and the onset of second-phase insulin release. One possible explanation for this discrepancy is that Rac1 has a role in perpetuating, rather than initiating, second-phase insulin release. A potential mechanism for this putative perpetuating effect may involve Rac1 signaling to Gelsolin.66, 106, 107 Rac1-dependent PIP2 production was suggested to be a driving force to inactivate Gelsolin, which in turn would alter the rate and/or localization of cortical F-actin remodeling.43 The development of specific acute inhibitors of Rac1 activation, as has been done recently for Cdc42,81, 108 should assist future studies to parse out the role of Rac1 signaling in actin remodeling and its relationship to second-phase insulin release in the β cell.
Other small GTPases with potential roles in β cell F-actin remodeling
Other small GTPases have been investigated for their roles in biphasic insulin secretion. For example, Arf6 was recently reported to become activated within the first minute of glucose stimulation and was proposed to be an upstream activator of the Cdc42 pathway.70 However, that Arf6 can be activated by KCl70 is inconsistent with the glucose-selective activation of Cdc42 and of second-phase insulin secretion,25 calling into question the role of Arf6 in the Cdc42–PAK1 signaling pathway. Another GTPase, Rap1, has also been shown to function as a positive regulator of insulin secretion. Rap1 is activated downstream of GLP-1 signaling in β cells due to cAMP-dependent activation of the Rap1 guanine nucleotide effector Epac2.75, 109 Rap1 is required for the full potentiation of glucose-induced insulin secretion by cAMP,75 but whether this is linked to F-actin remodeling is unknown. GTPases Rab27a21 and Rab3A22 are two additional positive effectors for both phases of insulin secretion.73 Although it is unclear if Rab3A is related to actin remodeling, Rab27a is implicated, as it signals downstream to myosin Va to promote insulin granule transport to the plasma membrane.110, 111
Role of ERK in insulin secretion and actin remodeling
More than 15 years ago, ERK was shown to be activated in response to glucose in β cells.112, 113 Although early studies concluded that active ERK translocated to the nucleus to exert effects upon transcription of insulin and other targets, a significant pool of cytoplasmic active ERK remained,96, 112, 113 and was later revealed to be necessary for glucose-stimulated insulin secretion in rat islets.96 In 2010, it was confirmed via RNAi-mediated depletion studies that ERK was indeed essential for glucose-stimulated insulin secretion, and solidified as an upstream activator of Rac1.105 Substantiating this signaling role for ERK, very recent work has suggested ERK may be important for amplification of insulin secretion in response to glucose.81, 114
The Ras–Raf–MEK–ERK pathway in the β cell
In the β cell, ERK has been shown to be activated in multiple ways. For example, glucose triggers calcium influx, calcium activates calcineurin to dephosphorylate B-Raf,115 and Raf then phosphorylates the MAP/ERK kinase (MEK) to subsequently phosphorylate ERK.116 Alternatively, Raf-1 was recently shown to be dependent upon Cdc42 activation and PAK1 signaling, which ultimately led to ERK activation.81 Early studies using pharmacological inhibition of ERK yielded conflicting results about its role in the β cell. Specifically, while inhibition of ERK activity had negligible impact upon stress-fiber actin remodeling,106 it did impact focal adhesion remodeling.14 More recent studies using a newer and more specific inhibitor support a role for ERK in glucose-stimulated cortical actin remodeling, with ERK positioned downstream of PAK1 signaling.81, 114 Thus, whether glucose-stimulated ERK activation in the β cell occurs in one sequential pathway downstream of Raf-1→PAK1, or instead is triggered concurrently via Ca2+→calcineurin→B-Raf, will require further investigation.
ERK targets actin regulatory proteins
ERK activation is clearly coupled to the actin cytoskeleton. ERK has been shown to phosphorylate synapsin I, cortactin, myosin light-chain kinase (MLCK), FAK and paxillin in other cell types. Although synapsin I does not have an essential role in glucose-stimulated insulin secretion,117 FAK and MLCK have been established as necessary factors;13, 118 cortactin is yet to be investigated in β cells. As FAK is required in β cells for basal and stimulated insulin secretion and focal adhesion remodeling,12, 13, 14 there exists the potential for input from the ERK pathway through FAK signaling to impact F-actin remodeling (Figure 3). ERK can also phosphorylate FAK to disrupt FAK-paxillin binding,119, 120, 121 and paxillin was suggested to be important for glucose-stimulated insulin secretion from primary rat β cells.14 MLCK has also been implicated in cortical F-actin remodeling and glucose-stimulated insulin secretion,103, 122 although its placement downstream of ERK in β cells remains to be demonstrated. Details of MLCK and FAK signaling in the β cell will be discussed in the following sections.
Actin-binding proteins in insulin granule exocytosis
As cortical F-actin remodeling is known to couple granule mobilization to the SNARE exocytosis machinery in a dynamic and transient manner,9, 12, 59, 66, 89 recent efforts have focused upon analyses of proteins that control the temporal and spatial polymerization and depolymerization events. The idea that F-actin severing and stabilizing proteins may underlie glucose-stimulated F-actin remodeling fits with the current model and also leaves room for the potential modifiers of F-actin dynamics in the β cell.
F-actin severing proteins in the β cell
β cells are known to express the F-actin severing proteins Gelsolin, Scinderin and Cofilin. Gelsolin is the founder of a family of calcium-activated actin-severing and -capping proteins, first cloned in 1979.123, 124 This protein family also includes Scinderin, CapG and many others,124 although only Gelsolin and Scinderin are known to be expressed in β cells. Gelsolin consists of six globular domains (S1–6), each of which binds calcium; S2 also binds to phosphoinositides.124 There are two isoforms of Gelsolin encoded as different mRNAs from the same gene: an intracellular cytosolic isoform and a secreted plasma isoform, which contains a 50-residue N-terminal extension.125, 126 The intracellular cytosolic form of Gelsolin participates in F-actin remodeling. Gelsolin was first implicated in glucose-stimulated insulin release in 2006.106 In 2012, a role of Gelsolin was determined to be in clamping unsolicited basal secretion, through its interaction with the target-SNARE protein, Syntaxin 4, at the plasma membrane66 (Figure 3). Scinderin shares nearly 60% sequence identity to Gelsolin,127 yet unlike Gelsolin, Scinderin expression is largely restricted to neuroendocrine cells (including islet β cells),128, 129 and is differentially regulated by calcium. Scinderin becomes activated at lower levels of calcium (0.5 μM) than Gelsolin (∼1–3 μM).130 In patch-clamped primary β cells, peptides derived from Scinderin’s PIP2-binding domain were shown to inhibit both calcium- and GTPγS-induced insulin secretion, potentially implicating Scinderin in glucose-stimulated insulin secretion.131 However, the role of Scinderin in glucose-induced F-actin remodeling and biphasic insulin release remains untested in the islet β cell. The least well-studied severing protein present in β cells is Cofilin. Cofilin is usually thought to be regulated upstream by PAK1 activation of LIM kinase. Active LIM kinase then phosphorylates Cofilin to inhibit its severing activity.132 Unexpectedly however, PAK1 deficient mouse islets and MIN6 β cells failed to show alterations in Cofilin activation, arguing against a significant role for Cofilin in glucose-induced F-actin remodeling and insulin release.27
F-actin stabilizing proteins with potential roles in the β cell
The two major F-actin stabilizing proteins, which may have potential roles in β cell F-actin remodeling, are α-Fodrin and Filamin A. α-Fodrin, also known as α-spectrin, cross-links F-actin under low calcium and releases actin upon calcium influx in chromaffin cells.52 α-Fodrin can be cleaved by Calpain and increased cleaved α-Fodrin is found in islet β cells from type 2 diabetic donors, suggesting compromised cellular cytoskeletal structure.133 α-Fodrin also binds to Syntaxin 4, the target-SNARE isoform operational in second-phase insulin release, in vitro.134, 135 Filamin A is the most potent of all known F-actin cross-linking proteins and interacts with Cdc42, PAK1 and Rac1, all of which are vital for second-phase insulin secretion.25, 136, 137, 138 PAK1 is known to phosphorylate Filamin A, regulating its F-actin cross-linking activity,99 although this is yet to be shown in β cells.
F-actin-associated proteins in the β cell
In addition to F-actin severing and stabilizing proteins, additional F-actin-associated proteins such as MLCK, myosin IIA and FAK, have been implicated in insulin release. In 1997, MLCK signaling to MLC2 was first implicated in glucose-stimulated insulin secretion,118 and glucose found to phosphorylate the myosin light-chain 2 (MLC2) subunit of myosin IIA at serine 19 in 1999.139 Myosin IIA is the most studied non-muscle myosin and is involved in F-actin remodeling and exocytosis.140, 141 In 2013, myosin IIA specifically, and not myosin IIB, was shown in primary β cells to be required for glucose-stimulated insulin secretion and actin remodeling.103 Whether MLCK→MLC2 requires upstream ERK activation in β cell cortical F-actin remodeling remains in question. This is because in β cells, ERK can also lie downstream of MLCK in the process of focal adhesion remodeling.103 In addition to myosin IIA, myosin Va has been shown to be a positive regulator of insulin granule localization to the plasma membrane and of glucose-stimulated insulin secretion.58 As such, it will be important to discern the ordered signaling events in cortical versus focal adhesion actin remodeling processes to reconcile these discordant data.
FAK regulation of focal adhesion remodeling has implications for indirect regulation of F-actin remodeling and insulin secretion. In MIN6 β cells, FAK inhibition was shown to prevent glucose-stimulated F-actin remodeling,12 decreasing glucose-stimulated insulin secretion.14 Recently, islets of β cell-specific FAK knockout mice were found to exhibit impaired glucose-induced F-actin reorganization correlated with decreased ERK activation, coinciding with a reduced number of docked insulin granules; serum insulin levels during glucose tolerance tests suggest that both phases of insulin secretion are decreased.13 Thus, FAK likely acts in coordination with F-actin remodeling in order to regulate glucose-stimulated insulin secretion, but its linkage to Cdc42 and PAK1 remains to be elucidated. Lastly, N-WASP and Arp2/3 proteins are known to cooperatively promote actin filament polymerization and are implicated in secretion from neuroendocrine cells,37 yet neither has been established in glucose-stimulated insulin secretion.
Snare-mediated insulin exocytosis
The concept of SNARE protein-mediated vesicle/granule exocytosis was first elucidated in 1993 and rapidly confirmed by multiple groups and in many diverse cell types, including β cells.142, 143, 144, 145, 146, 147 In β cells, the target-SNARE proteins, Syntaxin and synaptosomal-associated protein of 25 kDa (SNAP-25, as well as its homolog SNAP-23), are localized to the plasma membrane, whereas the vesicle-SNARE VAMP2 (also known as synaptobrevin 2) is localized to the insulin granule. Syntaxins have three N-terminal α-helices denoted Ha, Hb and Hc (Habc) followed by a linker domain and then the H3 SNARE helix domain that associates with the other SNARE proteins.145 VAMP2 and Syntaxin each donate their one alpha-helix and SNAP-23/25 contributes two alpha-helices to form a coiled four α-helical heterotrimeric complex that brings the plasma membrane and granule membranes in close enough proximity for fusion to occur;148 once fused, the insulin cargo is released from the β cell (Figure 3). SNARE complex formation is ATP dependent and the final initiation of regulated exocytosis requires calcium influx,149 although the exact mechanisms by which this calcium influx causes membrane fusion are not fully understood. In addition, other accessory factors are important for SNARE-mediated membrane fusion such as the Munc13 proteins and Doc2β, which also have calcium-binding activity that could aid in sensing increased cytosolic calcium.150 F-actin interacts with multiple SNARE proteins and can directly regulate exocytosis in this manner, therefore the linkage of F-actin and SNARE-mediated exocytosis will be discussed following a brief review of the role of SNAREs in biphasic insulin secretion.
SNARE requirement in biphasic insulin secretion
In β cells, the necessary SNARE isoforms for glucose-stimulated insulin secretion are Syntaxin 1,23 Syntaxin 3,151, 152 Syntaxin 4,17 SNAP-25,153 SNAP-23154 and VAMP2.155 Indeed, islets from type 2 diabetic donors have low abundances of Syntaxins 1 and 4, SNAP-25 and VAMP2, highlighting the functional importance of these proteins in the disease state (unpublished data, E. Oh and D.C. Thurmond).156, 157 Syntaxin 1 is required for first-phase insulin secretion only, while Syntaxin 4 has been shown as necessary for both phases of insulin secretion.17, 23 Most recently, a third isoform, Syntaxin 3, has been implicated in granule recruitment to support both phases of glucose-stimulated insulin secretion.151 Syntaxin protein function is tightly regulated by binding to their cognate nSec1/Munc18 (SM) proteins. Syntaxin 1 and 3 are regulated by Munc18-1,158 Syntaxin 3 may also be regulated by Munc18-2 (also known as Munc18b) in β cells.152 Distinct from these isoforms is the pairing of Syntaxin 4 with Munc18c (also known as Munc18-3) with Munc18c being the only isoform exclusively required for the second phase of insulin secretion.28 It is hypothesized that upon stimulation, post-translational modification of SM proteins causes dissociation (in the case of Munc18c) or a binding mode shift (in the case of Munc18-1) to assist in the opening and binding of Syntaxin to its cognate vesicle-SNARE VAMP2.150 Post-translational modification of the Syntaxin proteins also contributes to their activity, as glucose-induced production of nitric oxide causes S-nitrosylation of Syntaxin 4 in β cells.159 Most recently, Munc13-4, the newest member of the Munc13 family of priming factors, was shown to interact with the H3 SNARE domains of Syntaxins 1 and 4 in a calcium-dependent manner.160 Munc13-4 also interacts with the small GTPase Rab27a,161 and therefore could be a missing link coupling glucose-regulated GTPases to SNARE-mediated biphasic insulin exocytosis.
Linkage of F-actin remodeling to SNARE-mediated secretion
F-actin function is mechanistically coupled to multiple SNARE and SNARE-associated proteins and many F-actin-binding proteins interact with the SNARE machinery. Immunoprecipitation of Syntaxin 1, Syntaxin 4 and SNAP-25 from β cells show co-precipitation of F-actin.9, 12, 59, 66, 162 Of interest is the dynamic nature of the F-actin-SNARE interaction after glucose stimulation. After 5–10 min of glucose stimulation, F-actin dissociates from the Syntaxins and SNAP-25, but re-associates within 30 min after stimulation.9, 12, 59 In the case of Syntaxin 4, this binding has been shown to be direct.9, 34, 59, 162 Direct binding of F-actin to Syntaxin 4 was determined to occur at the N-terminus of Syntaxin 4, likely through the Hb helix.59 The Syntaxin 4–F-actin interaction seems to be selective for Syntaxin 4, as all other SNARE proteins tested so far (Syntaxins 1, 2 and 3, SNAP-23, VAMP2 and VAMP8) have not bound F-actin directly in vitro.44, 59, 162 In β cells, evidence for Syntaxin 4 binding to F-actin and not G-actin comes from treatment with the actin depolymerizing drug latrunculin, which abrogates the co-precipitation of actin with Syntaxin 4, showing that Syntaxin 4 will not bind to monomeric G-actin.59, 66 Artificial disruption of Syntaxin 4–F-actin complexes in β cells using a GFP-tagged truncation of Syntaxin 4 corresponding to the F-actin-binding site led to increased insulin secretion and VAMP2-bound insulin granule accumulation at the cell surface, suggesting that F-actin binding to Syntaxin 4 has a role in restricting granule access to the plasma membrane in the basal or unstimulated state.59 Syntaxin 1 and 4 can also form heterotrimeric complexes with Cdc42 and VAMP2 in vitro, presenting a potential avenue for localized coupling of SNARE proteins with Cdc42-mediated F-actin remodeling.89, 163
Concluding remarks
What mediates the glucose-specific signal for β cell F-actin remodeling? Glucose metabolism results in calcium influx required to trigger insulin secretion, and it is now recognized that a metabolic amplifying signal/second messenger is also produced, although the identity of this amplifying factor has remained elusive.164, 165 Two recent studies used the rat β cell line INS-1 832/13 to investigate glucose-induced changes in different metabolites.166, 167 These studies implicated the pentose phosphate pathway as well as AMPK kinase activation (from ZMP) as metabolites with the potential to be the glucose sensing readout that leads to insulin secretion. Future work with such metabolites may lead to the discovery of the signal that leads to activation of the Cdc42–PAK1–Rac1 pathway that is critical for F-actin remodeling and second-phase insulin secretion.
F-actin remodeling has been investigated for a role in the amplification of insulin secretion. Recently, the diazoxide paradigm was used in islets in conjunction with pharmacological disruption of F-actin using latrunculin or stabilization of F-actin using jasplakinolide.10 Polymerization or depolymerization of F-actin elicited potentiation of biphasic insulin secretion, consistent with the published literature.9, 10 These studies highlight how F-actin remodeling may fine-tune and regulate the amount of insulin that is secreted in response to the triggering and amplifying mechanisms. A caveat to the use of pharmacological de/polymerizing agents complicates determination of the role of F-actin, however, as pre-treatment with these drugs alters the F/G-actin ratio and granule distribution at the plasma membrane prior to stimulation, and they cause global changes to F-actin, as opposed to the localized alterations that are seen to occur in response to glucose.9, 34 As such, it is important to study F-actin remodeling in β cells using reagents that can act more selectively, and in a localized manner, upon key regulators of F-actin dynamics in order to elucidate more nuanced regulatory roles of localized cytoskeletal reorganization. Future studies incorporating novel tools in the β cell, such as light-activated small Rho GTPases, will be instrumental in addressing the role of localized F-actin remodeling on insulin exocytosis.168, 169, 170
References
Ashcroft FM, Harrison DE, Ashcroft SJ . Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984; 312: 446–448.
Cook DL, Hales CN . Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984; 311: 271–273.
Rorsman P, Ashcroft FM, Trube G . Single Ca channel currents in mouse pancreatic B-cells. Pflugers Arch 1988; 412: 597–603.
Satin LS, Cook DL . Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Arch 1985; 404: 385–387.
Grodsky GM . Kinetics of insulin secretion: underlying metabolic events. In: LeRoith D, Taylor S, Olefsky J (eds). Diabetes Mellitus: a fundamental and clinical text. Lippincott Williams & Wilkins: Philadelphia, PA, 2000.
Daniel S, Noda M, Straub SG, Sharp GW, Komatsu M, Schermerhorn T et al. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes 1999; 48: 1686–1690.
Rorsman P, Eliasson L, Renstrom E, Gromada J, Barg S, Gopel S . The cell physiology of biphasic insulin secretion. News Physiol Sci 2000; 15: 72–77.
Rorsman P, Braun M . Regulation of insulin secretion in human pancreatic islets. Ann Rev Physiol 2013; 75: 155–179.
Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE . Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol Endocrinol 2003; 17: 732–742.
Mourad NI, Nenquin M, Henquin JC . Metabolic amplifying pathway increases both phases of insulin secretion independently of beta-cell actin microfilaments. Am J Physiol Cell Physiol 2010; 299: C389–C398.
Wang Z, Thurmond DC . Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 2009; 122: 893–903.
Rondas D, Tomas A, Soto-Ribeiro M, Wehrle-Haller B, Halban PA . Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J Biol Chem 2012; 287: 2423–2436.
Cai EP, Casimir M, Schroer SA, Luk CT, Shi SY, Choi D et al. In Vivo Role of focal adhesion kinase in regulating pancreatic beta-cell mass and function through insulin signaling, actin dynamics, and granule trafficking. Diabetes 2012; 61: 1708–1718.
Rondas D, Tomas A, Halban PA . Focal adhesion remodeling is crucial for glucose-stimulated insulin secretion and involves activation of focal adhesion kinase and paxillin. Diabetes 2011; 60: 1146–1157.
Nesher R, Cerasi E . Modeling phasic insulin release: immediate and time-dependent effects of glucose. Diabetes 2002; 51: S53–S59.
Gerich JE . Is reduced first-phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes? Diabetes 2002; 51: S117–S121.
Spurlin BA, Thurmond DC . Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic {beta}-cells. Mol Endocrinol 2006; 20: 183–193.
Gomi H, Mizutani S, Kasai K, Itohara S, Izumi T . Granuphilin molecularly docks insulin granules to the fusion machinery. J Cell Biol 2005; 171: 99–109.
Bhatnagar S, Oler AT, Rabaglia ME, Stapleton DS, Schueler KL, Truchan NA et al. Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet 2011; 7: e1002323.
Lopez JA, Kwan EP, Xie L, He Y, James DE, Gaisano HY . The RalA GTPase is a central regulator of insulin exocytosis from pancreatic islet beta cells. J Biol Chem 2008; 283: 17939–17945.
Kasai K, Ohara-Imaizumi M, Takahashi N, Mizutani S, Zhao S, Kikuta T et al. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 2005; 115: 388–396.
Yaekura K, Julyan R, Wicksteed BL, Hays LB, Alarcon C, Sommers S et al. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem 2003; 278: 9715–9721.
Ohara-Imaizumi M, Fujiwara T, Nakamichi Y, Okamura T, Akimoto Y, Kawai J et al. Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis. J Cell Biol 2007; 177: 695–705.
Ferdaoussi M, Bergeron V, Zarrouki B, Kolic J, Cantley J, Fielitz J et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 2012; 55: 2682–2692.
Wang Z, Oh E, Thurmond DC . Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem 2007; 282: 9536–9546.
Wang Z, Thurmond DC . Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J Biol Chem 2010; 285: 6186–6197.
Wang Z, Oh E, Clapp DW, Chernoff J, Thurmond DC . Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J Biol Chem 2011; 286: 41359–41367.
Oh E, Thurmond DC . Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes 2009; 58: 1165–1174.
Orci L, Gabbay KH, Malaisse WJ . Pancreatic beta-cell web: its possible role in insulin secretion. Science 1972; 175: 1128–1130.
Aunis D, Bader MF . The cytoskeleton as a barrier to exocytosis in secretory cells. J Exp Biol 1988; 139: 253–266.
Stutchfield J, Howell SL . The effect of phalloidin on insulin secretion from islets of Langerhans isolated from rat pancreas. FEBS Lett 1984; 175: 393–396.
van Obberghen E, Somers G, Devis G, Vaughan GD, Malaisse-Lagae F, Orci L et al. Dynamics of insulin release and microtubular-microfilamentous system. I. Effect of cytochalasin B. J Clin Invest 1973; 52: 1041–1051.
Li G, Rungger-Brandle E, Just I, Jonas JC, Aktories K, Wollheim CB . Effect of disruption of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Mol Biol Cell 1994; 5: 1199–1213.
Nevins AK, Thurmond DC . Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol 2003; 285: C698–C710.
Jerdeva GV, Wu K, Yarber FA, Rhodes CJ, Kalman D, Schechter JE et al. Actin and non-muscle myosin II facilitate apical exocytosis of tear proteins in rabbit lacrimal acinar epithelial cells. J Cell Sci 2005; 118: 4797–4812.
Lim GE, Xu M, Sun J, Jin T, Brubaker PL . The Rho Guanosine 5'-triphosphatase, cell division cycle 42, is required for insulin-induced actin remodeling and glucagon-like peptide-1 secretion in the intestinal endocrine L cell. Endocrinology 2009; 150: 580–591.
Gasman S, Chasserot-Golaz S, Malacombe M, Way M, Bader MF . Regulated exocytosis in neuroendocrine cells: a role for subplasmalemmal Cdc42/N-WASP-induced actin filaments. Mol Biol Cell 2004; 15: 520–531.
Gil A, Rueda J, Viniegra S, Gutierrez LM . The F-actin cytoskeleton modulates slow secretory components rather than readily releasable vesicle pools in bovine chromaffin cells. Neuroscience 2000; 98: 605–614.
Chowdhury HH, Popoff MR, Zorec R . Actin cytoskeleton depolymerization with clostridium spiroforme toxin enhances the secretory activity of rat melanotrophs. J Physiol 1999; 521 (Pt 2): 389–395.
Carbajal ME, Vitale ML . The cortical actin cytoskeleton of lactotropes as an intracellular target for the control of prolactin secretion. Endocrinology 1997; 138: 5374–5384.
Pendleton A, Koffer A . Effects of latrunculin reveal requirements for the actin cytoskeleton during secretion from mast cells. Cell Motil Cytoskeleton 2001; 48: 37–51.
Johnson JL, Monfregola J, Napolitano G, Kiosses WB, Catz SD . Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effector JFC1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol Biol Cell 2012; 23: 1902–1916.
Azuma T, Witke W, Stossel TP, Hartwig JH, Kwiatkowski DJ . Gelsolin is a downstream effector of rac for fibroblast motility. EMBO J 1998; 17: 1362–1370.
Woronowicz K, Dilks JR, Rozenvayn N, Dowal L, Blair PS, Peters CG et al. The platelet actin cytoskeleton associates with SNAREs and participates in alpha-granule secretion. Biochemistry 2010; 49: 4533–4542.
Wang F, Wang Y, Kim MS, Puthanveetil P, Ghosh S, Luciani DS et al. Glucose-induced endothelial heparanase secretion requires cortical and stress actin reorganization. Cardiovasc Res 2010; 87: 127–136.
Dillon C, Goda Y . The actin cytoskeleton: integrating form and function at the synapse. Ann Rev Neurosci 2005; 28: 25–55.
Omata W, Shibata H, Li L, Takata K, Kojima I . Actin filaments play a critical role in insulin-induced exocytotic recruitment but not in endocytosis of GLUT4 in isolated rat adipocytes. Biochem J 2000; 346: 321–328.
Brozinick JT Jr, Hawkins ED, Strawbridge AB, Elmendorf JS . Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J Biol Chem 2004; 279: 40699–40706.
Aunis D, Guerold B, Bader MF, Cieselski-Treska J . Immunocytochemical and biochemical demonstration of contractile proteins in chromaffin cells in culture. Neuroscience 1980; 5: 2261–2277.
Burgoyne RD, Cheek TR, Norman KM . Identification of a secretory granule-binding protein as caldesmon. Nature 1986; 319: 68–70.
Vitale ML, Rodriguez Del Castillo A, Tchakarov L, Trifaro JM . Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J Cell Biol 1991; 113: 1057–1067.
Perrin D, Aunis D . Reorganization of alpha-fodrin induced by stimulation in secretory cells. Nature 1985; 315: 589–592.
Somers G, Blondel B, Orci L, Malaisse WJ . Motile events in pancreatic endocrine cells. Endocrinology 1979; 104: 255–264.
Wilson JR, Ludowyke RI, Biden TJ . A redistribution of actin and myosin IIA accompanies Ca(2+)-dependent insulin secretion. FEBS Lett 2001; 492: 101–106.
Wang JL, Easom RA, Hughes JH, McDaniel ML . Evidence for a role of microfilaments in insulin release from purified beta-cells. Biochem Biophys Res Commun 1990; 171: 424–430.
Swanston-Flatt SK, Carlsson L, Gylfe E . Actin filament formation in pancreatic beta-cells during glucose stimulation of insulin secretion. FEBS Lett 1980; 117: 299–302.
Howell SL, Tyhurst M . Regulation of actin polymerizaton in rat islets of Langerhans. Biochem J 1980; 192: 381–383.
Varadi A, Tsuboi T, Rutter GA . Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol Biol Cell 2005; 16: 2670–2680.
Jewell JL, Luo W, Oh E, Wang Z, Thurmond DC . Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J Biol Chem 2008; 283: 10716–10726.
Schubert S, Knoch KP, Ouwendijk J, Mohammed S, Bodrov Y, Jager M et al. beta2-Syntrophin is a Cdk5 substrate that restrains the motility of insulin secretory granules. PloS One 2010; 5: e12929.
Snabes MC, Boyd AE . Increased filamentous actin in islets of Langerhans from fasted hamsters. Biochem Biophys Res Commun 1982; 104: 207–211.
Konstantinova I, Nikolova G, Ohara-Imaizumi M, Meda P, Kucera T, Zarbalis K et al. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 2007; 129: 359–370.
Corkey BE . Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes 2012; 61: 4–13.
Kepner EM, Yoder SM, Oh E, Kalwat MA, Wang Z, Quilliam LA et al. Cool-1/betaPIX functions as a guanine nucleotide exchange factor in the cycling of Cdc42 to regulate insulin secretion. Am J Physiol Endocrinol Metab 2011; 301: E1072–E1080.
Nevins AK, Thurmond DC . Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J Biol Chem 2006; 281: 18961–18972.
Kalwat MA, Wiseman DA, Luo W, Wang Z, Thurmond DC . Gelsolin associates with the N-terminus of Syntaxin 4 to regulate insulin granule exocytosis. Mol Endocrinol 2012; 26: 128–141.
Bishop AL, Hall A . Rho GTPases and their effector proteins. Biochem J 2000; 348: 241–255.
Wennerberg K, Rossman KL, Der CJ . The Ras superfamily at a glance. J Cell Sci 2005; 118: 843–846.
DerMardirossian C, Bokoch GM . GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005; 15: 356–363.
Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A . Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem Pharmacol 2011; 81: 1016–1027.
Brozzi F, Diraison F, Lajus S, Rajatileka S, Philips T, Regazzi R et al. Molecular mechanism of myosin Va recruitment to dense core secretory granules. Traffic 2012; 13: 54–69.
Wang H, Ishizaki R, Kobayashi E, Fujiwara T, Akagawa K, Izumi T . Loss of granuphilin and loss of syntaxin-1A cause differential effects on insulin granule docking and fusion. J Biol Chem 2011; 286: 32244–32250.
Merrins MJ, Stuenkel EL . Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J Physiol 2008; 586: 5367–5381.
Izumi T . Physiological roles of Rab27 effectors in regulated exocytosis. Endocr J. 2007; 54: 649–657.
Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333–19338.
Kowluru A, Li G, Rabaglia ME, Segu VB, Hofmann F, Aktories K et al. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cells. Biochem Pharmacol 1997; 54: 1097–1108.
Hammar E, Tomas A, Bosco D, Halban PA . Role of the Rho-ROCK (Rho-associated kinase) signaling pathway in the regulation of pancreatic beta-cell function. Endocrinology 2009; 150: 2072–2079.
Li J, Luo R, Kowluru A, Li G . Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am J Physiol Endocrinol Metab 2004; 286: E818–E827.
Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013; 56: 1088–1097.
Kesavan G, Sand FW, Greiner TU, Johansson JK, Kobberup S, Wu X et al. Cdc42-mediated tubulogenesis controls cell specification. Cell 2009; 139: 791–801.
Kalwat MA, Yoder SM, Wang Z, Thurmond DC. A . p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic beta cells. Biochem Pharmacol 2013; 85: 808–816.
Kowluru A . Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 2011; 81: 965–975.
Kowluru A, Veluthakal R . Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 2005; 54: 3523–3529.
Hansen CG, Nichols BJ . Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol 2010; 20: 177–186.
Thomas CM, Smart EJ . Caveolae structure and function. J Cell Mol Med 2008; 12: 796–809.
Ishikawa Y, Otsu K, Oshikawa J . Caveolin; different roles for insulin signal? Cell Signal 2005; 17: 1175–1182.
Cheng ZJ, Singh RD, Holicky EL, Wheatley CL, Marks DL, Pagano RE . Co-regulation of caveolar and Cdc42-dependent fluid phase endocytosis by phosphocaveolin-1. J Biol Chem 2010; 285: 15119–15125.
Baltierrez-Hoyos R, Roa-Espitia AL, Hernandez-Gonzalez EO . The association between CDC42 and caveolin-1 is involved in the regulation of capacitation and acrosome reaction of guinea pig and mouse sperm. Reproduction 2012; 144: 123–134.
Nevins AK, Thurmond DC . A direct interaction between Cdc42 and vesicle-associated membrane protein 2 regulates SNARE-dependent insulin exocytosis. J Biol Chem 2005; 280: 1944–1952.
Bokoch GM . Biology of the P21-Activated Kinases. Ann Rev Biochem 2003; 72: 743–781.
Finkielstein CV, Overduin M, Capelluto DG . Cell migration and signaling specificity is determined by the phosphatidylserine recognition motif of Rac1. J Biol Chem 2006; 281: 27317–27326.
Diebold BA, Fowler B, Lu J, Dinauer MC, Bokoch GM . Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J Biol Chem 2004; 279: 28136–28142.
Nie J, Sun C, Faruque O, Ye G, Li J, Liang Q et al. Synapses of amphids defective (SAD-A) kinase promotes glucose-stimulated insulin secretion through activation of p21-activated kinase (PAK1) in pancreatic beta-cells. J Biol Chem 2012; 287: 26435–26444.
Nie J, Lilley BN, Pan YA, Faruque O, Liu X, Zhang W et al. SAD-A potentiates glucose-stimulated insulin secretion as a mediator of glucagon-like peptide 1 response in pancreatic beta cells. Mol Cell Bio 2013; 33: 2527–2534.
Chen XY, Gu XT, Saiyin H, Wan B, Zhang YJ, Li J et al. Brain selective kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic beta-cells. J Biol Chem 2012; 287: 30368–30375.
Longuet C, Broca C, Costes S, Hani EH, Bataille D, Dalle S . Extracellularly regulated kinases 1/2 (p44/42 mitogen-activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 beta-cell line and islets of Langerhans. Endocrinology 2005; 146: 643–654.
Trumper J, Ross D, Jahr H, Brendel MD, Goke R, Horsch D . The Rap-B-Raf signalling pathway is activated by glucose and glucagon-like peptide-1 in human islet cells. Diabetologia 2005; 48: 1534–1540.
Edwards DC, Sanders LC, Bokoch GM, Gill GN . Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1999; 1: 253–259.
Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP et al. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol 2002; 4: 681–690.
Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P . Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999; 283: 2083–2085.
DerMardirossian C, Schnelzer A, Bokoch GM . Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell 2004; 15: 117–127.
Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R . p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO R 2004; 5: 154–160.
Arous C, Rondas D, Halban PA . Non-muscle myosin IIA is involved in focal adhesion and actin remodelling controlling glucose-stimulated insulin secretion. Diabetologia 2013; 56: 792–802.
Kowluru A . Small G proteins in islet beta-cell function. Endocr Rev 2010; 31: 52–78.
Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ . Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta-cells. Diabetes 2010; 59: 967–977.
Tomas A, Yermen B, Min L, Pessin JE, Halban PA . Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: role of gelsolin and cooperation with the MAPK signalling pathway. J Cell Sci 2006; 119: 2156–2167.
Casimir M, Dai XQ, Hajmrle C, Kolic J, Guo D, Oudit G et al. Gelsolin knockout impairs insulin secretion independently of actin polymerization. Diabetes 2011; 60: A351.
Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem 2013; 288: 8531–8543.
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998; 396: 474–477.
Mizuno K, Ramalho JS, Izumi T . Exophilin8 transiently clusters insulin granules at the actin-rich cell cortex prior to exocytosis. Mol Biol Cell 2011; 22: 1716–1726.
Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C et al. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell 2003; 14: 4103–4113.
Khoo S, Cobb MH . Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci USA 1997; 94: 5599–5604.
Frodin M, Sekine N, Roche E, Filloux C, Prentki M, Wollheim CB et al. Glucose, other secretagogues, and nerve growth factor stimulate mitogen-activated protein kinase in the insulin-secreting beta-cell line, INS-1. J Biol Chem 1995; 270: 7882–7889.
Bowe JE, Chander A, Liu B, Persaud SJ, Jones PM . The permissive effects of glucose on receptor-operated potentiation of insulin secretion from mouse islets: a role for ERK1/2 activation and cytoskeletal remodelling. Diabetologia 2013; 56: 783–791.
Duan L, Cobb MH . Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc Natl Acad Sci USA 2010; 107: 22314–22319.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–183.
Wendt A, Speidel D, Danielsson A, Esguerra JL, Bogen IL, Walaas SI et al. Synapsins I and II are not required for insulin secretion from mouse pancreatic beta-cells. Endocrinology 2012; 153: 2112–2119.
Iida Y, Senda T, Matsukawa Y, Onoda K, Miyazaki JI, Sakaguchi H et al. Myosin light-chain phosphorylation controls insulin secretion at a proximal step in the secretory cascade. Am J Physiol 1997; 273: E782–E789.
Hunger-Glaser I, Salazar EP, Sinnett-Smith J, Bombesin Rozengurt E . lysophosphatidic acid, and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: requirement for ERK activation. J Biol Chem 2003; 278: 22631–22643.
Dobkin-Bekman M, Naidich M, Rahamim L, Przedecki F, Almog T, Lim S et al. A preformed signaling complex mediates GnRH-activated ERK phosphorylation of paxillin and FAK at focal adhesions in L beta T2 gonadotrope cells. Mol Endocrinol 2009; 23: 1850–1864.
Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell 2009; 35: 11–25.
Yu W, Niwa T, Fukasawa T, Hidaka H, Senda T, Sasaki Y et al. Synergism of protein kinase A, protein kinase C, and myosin light-chain kinase in the secretory cascade of the pancreatic beta-cell. Diabetes 2000; 49: 945–952.
Yin HL, Stossel TP . Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature 1979; 281: 583–586.
Kwiatkowski DJ . Functions of gelsolin: motility, signaling, apoptosis, cancer. Curr Opin Cell Biol. 1999; 11: 103–108.
Yin HL, Kwiatkowski DJ, Mole JE, Cole FS . Structure and biosynthesis of cytoplasmic and secreted variants of gelsolin. J Biol Chem 1984; 259: 5271–5276.
Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL . Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature 1986; 323: 455–458.
Bader MF, Trifaro JM, Langley OK, Thierse D, Aunis D . Secretory cell actin-binding proteins: identification of a gelsolin-like protein in chromaffin cells. J Cell Biol 1986; 102: 636–646.
Lueck A, Brown D, Kwiatkowski DJ . The actin-binding proteins adseverin and gelsolin are both highly expressed but differentially localized in kidney and intestine. J Cell Sci 1998; 111 (Pt 24): 3633–3643.
Tchakarov L, Vitale ML, Jeyapragasan M, Rodriguez Del Castillo A, Trifaro JM . Expression of scinderin, an actin filament-severing protein, in different tissues. FEBS Lett 1990; 268: 209–212.
Lueck A, Yin HL, Kwiatkowski DJ, Allen PG . Calcium regulation of gelsolin and adseverin: a natural test of the helix latch hypothesis. Biochemistry 2000; 39: 5274–5279.
Bruun TZ, Hoy M, Gromada J . Scinderin-derived actin-binding peptides inhibit Ca(2+)- and GTPgammaS-dependent exocytosis in mouse pancreatic beta-cells. Eur J Pharmacol 2000; 403: 221–224.
Mizuno K . Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal 2013; 25: 457–469.
Huang CJ, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S et al. Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem 2010; 285: 339–348.
Nakano M, Nogami S, Sato S, Terano A, Shirataki H . Interaction of syntaxin with alpha-fodrin, a major component of the submembranous cytoskeleton. Biochem Biophys Res Commun 2001; 288: 468–475.
Liu L, Jedrychowski MP, Gygi SP, Pilch PF . Role of insulin-dependent cortical fodrin/spectrin remodeling in glucose transporter 4 translocation in rat adipocytes. Mol Biol Cell 2006; 17: 4249–4256.
Ito T, Suzuki A, Stossel TP . Regulation of water flow by actin-binding protein-induced actin gelatin. Biophys J. 1992; 61: 1301–1305.
Popowicz GM, Schleicher M, Noegel AA, Holak TA . Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci. 2006; 31: 411–419.
Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2001; 2: 138–145.
Wilson JR, Biden TJ, Ludowyke RI . Increases in phosphorylation of the myosin II heavy chain, but not regulatory light chains, correlate with insulin secretion in rat pancreatic islets and RINm5F cells. Diabetes 1999; 48: 2383–2389.
Tan JL, Ravid S, Spudich JA . Control of nonmuscle myosins by phosphorylation. Ann Rev Biochem 1992; 61: 721–759.
Andzelm MM, Chen X, Krzewski K, Orange JS, Strominger JL . Myosin IIA is required for cytolytic granule exocytosis in human NK cells. J Exp Med 2007; 204: 2285–2291.
Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P et al. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993; 362: 318–324.
Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE . A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993; 75: 409–418.
Rizo J, Sudhof TC . Snares and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci 2002; 3: 641–653.
Chen YA, Scheller RH . SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2001; 2: 98–106.
Scales SJ, Chen YA, Yoo BY, Patel SM, Doung YC, Scheller RH . SNAREs contribute to the specificity of membrane fusion. Neuron 2000; 26: 457–464.
Wheeler MB, Sheu L, Ghai M, Bouquillon A, Grondin G, Weller U et al. Characterization of SNARE protein expression in beta cell lines and pancreatic islets. Endocrinology 1996; 137: 1340–1348.
Jahn R, Scheller RH . SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol 2006; 7: 631–643.
Eliasson L, Renstrom E, Ding WG, Proks P, Rorsman P . Rapid ATP-dependent priming of secretory granules precedes Ca(2+)-induced exocytosis in mouse pancreatic B-cells. J Physiol 1997; 503: 399–412.
Jewell JL, Oh E, Thurmond DC . Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. AJP Regul Integr Comp Physiol 2010; 298: R517–R531.
Zhu D, Koo E, Kwan E, Kang Y, Park S, Xie H et al. Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia 2013; 56: 359–369.
Lam PP, Ohno M, Dolai S, He Y, Qin T, Liang T et al. Munc18b is a major mediator of insulin exocytosis in rat pancreatic beta-cells. Diabetes 2013; 62 (7): 2416–2428.
Sadoul K, Lang J, Montecucco C, Weller U, Regazzi R, Catsicas S et al. SNAP-25 is expressed in islets of Langerhans and is involved in insulin release. J Cell Biol 1995; 128: 1019–1028.
Sadoul K, Berger A, Niemann H, Weller U, Roche PA, Klip A et al. SNAP-23 is not cleaved by botulinum neurotoxin E and can replace SNAP-25 in the process of insulin secretion. J Biol Chem 1997; 272: 33023–33027.
Regazzi R, Wollheim CB, Lang J, Theler JM, Rossetto O, Montecucco C et al. VAMP-2 and cellubrevin are expressed in pancreatic beta-cells and are essential for Ca(2+)-but not for GTP gamma S-induced insulin secretion. EMBO J 1995; 14: 2723–2730.
Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T . Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 2006; 55: 435–440.
Andersson SA, Olsson AH, Esguerra JL, Heimann E, Ladenvall C, Edlund A et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol Cell Endocrinol 2012; 364: 36–45.
Hata Y, Slaughter CA, Sudhof TC . Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature 1993; 366: 347–351.
Wiseman DA, Kalwat MA, Thurmond DC . Stimulus-induced S-nitrosylation of syntaxin 4 impacts insulin granule exocytosis. J Biol Chem 2011; 286: 16344–16354.
Boswell KL, James DJ, Esquibel JM, Bruinsma S, Shirakawa R, Horiuchi H et al. Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J Cell Biol 2012; 197: 301–312.
Shirakawa R, Higashi T, Tabuchi A, Yoshioka A, Nishioka H, Fukuda M et al. Munc13-4 is a GTP-Rab27-binding protein regulating dense core granule secretion in platelets. J Biol Chem 2004; 279: 10730–10737.
Band AM, Ali H, Vartiainen MK, Welti S, Lappalainen P, Olkkonen VM et al. Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton. FEBS Lett 2002; 531: 513–519.
Daniel S, Noda M, Cerione RA, Sharp GW . A link between Cdc42 and syntaxin is involved in mastoparan-stimulated insulin release. Biochemistry 2002; 41: 9663–9671.
Henquin JC . Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 2009; 52: 739–751.
Henquin JC . Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000; 49: 1751–1760.
Lorenz MA, El Azzouny MA, Kennedy RT, Burant CF . Metabolome Response to Glucose in the beta-Cell Line INS-1 832/13. J Biol Chem 2013; 288: 10923–10935.
Spegel P, Sharoyko VV, Goehring I, Danielsson AP, Malmgren S, Nagorny CL et al. Time-resolved metabolomics analysis of beta-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem J 2013; 450: 595–605.
DeRose R, Pohlmeyer C, Umeda N, Ueno T, Nagano T, Kuo S et al. Spatio-temporal manipulation of small GTPase activity at subcellular level and on timescale of seconds in living cells. J Vis Exp 2012; 61 e 3794.
Wu YI, Wang X, He L, Montell D, Hahn KM . Spatiotemporal control of small GTPases with light using the LOV domain. Methods Enzymol 2011; 497: 393–407.
Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009; 461: 104–108.
Acknowledgements
This study was supported by grants from the National Institutes of Health (DK067912 and DK076614 to DCT) and the American Heart Association (10PRE3040010 to MAK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kalwat, M., Thurmond, D. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp Mol Med 45, e37 (2013). https://doi.org/10.1038/emm.2013.73
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.73
Keywords
This article is cited by
-
Selective serotonin reuptake inhibitor, fluoxetine, impairs E-cadherin-mediated cell adhesion and alters calcium homeostasis in pancreatic beta cells
Scientific Reports (2017)
-
Targeting insulin-producing beta cells for regenerative therapy
Diabetologia (2016)
-
Yap1 plays a protective role in suppressing free fatty acid-induced apoptosis and promoting beta-cell survival
Protein & Cell (2016)
-
Effect of Cell Adhesion Molecule 1 Expression on Intracellular Granule Movement in Pancreatic α Cells
Cell Biochemistry and Biophysics (2016)
-
Identification of proliferative and mature β-cells in the islets of Langerhans
Nature (2016)
