
Abstract
TNF-α is a major cytokine involved in inflammatory bowel disease (IBD). In this study, water extract of Grifola frondosa (GFW) was evaluated for its protective effects against colon inflammation through the modulation of TNF-α action. In coculture of HT-29 human colon cancer cells with U937 human monocytic cells, TNF-α-induced monocyte adhesion to HT-29 cells was significantly suppressed by GFW (10, 50, 100 µg/ml). The reduced adhesion by GFW correlated with the suppressed expression of MCP-1 and IL-8, the major IBD-associated chemokines. In addition, treatment with GFW significantly suppressed TNF-α-induced reactive oxygen species production and NF-κB transcriptional activity in HT-29 cells. In differentiated U937 monocytic cells, LPS-induced TNF-α production, which is known to be mediated through NF-κB activation, was significantly suppressed by GFW. In an in vivo rat model of IBD, oral administration of GFW for 5 days (1 g/kg per day) significantly inhibited the trinitrobenzene sulfonic acid (TNBS)-induced weight loss, colon ulceration, myeloperoxidase activity, and TNF-α expression in the colon tissue. Moreover, the effect of GFW was similar to that of intra-peritoneal injection of 5-aminosalicylic acid (5-ASA), an active metabolite of sulfasalazine, commonly used drug for the treatment of IBD. The results suggest that GFW ameliorates colon inflammation by suppressing production of TNF-α as well as its signaling through NF-κB leading to the expression of inflammatory chemokines, MCP-1 and IL-8. Taken together, the results strongly suggest GFW is a valuable medicinal food for IBD treatment, and thus may be used as an alternative medicine for IBD.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD) encompasses two chronic intestinal diseases, Crohn's disease (CD) and ulcerative colitis (UC), which are characterized by recurrent flare of inflammation in the gastrointestinal tract (Podolsky, 2002; Elson et al., 2005). During recurrent inflammatory process, activated and infiltrated leukocytes produce pro-inflammatory cytokines, such as TNF-α. Early studies on IBD have shown that TNF-α expression is increased in blood (Komatsu et al., 2001), intestinal mucosa (Murch et al., 1993; Autschbach et al., 1995), stools (Braegger et al., 1992), and cultured intestinal biopsies from IBD patients (Reimund et al., 1996), suggesting that TNF-α plays a critical role in intestinal inflammation (Murch et al., 1993). Upon stimulation with TNF-α, intestinal epithelium produces reactive oxygen species (ROS) through NAD(P)H oxidase (Nox) activation (Kim et al., 2007; Babu et al., 2008). Such TNF-α-induced ROS leads to activation of the transcription factor, NF-κB, which is a major regulator of inflammatory gene expression (Wajant et al., 2003). Based on its pleiotropic pro-inflammatory nature, anti-TNF-α antibody has been developed for the treatment of IBD refractory to standard treatment with steroids (Targan et al., 1997; Present et al., 1999). However, there are some limitations in anti-TNF-α antibody therapy. Some patients develop side effects such as infusion reactions or infectious complications in addition to the high costs of this therapy (Keane et al., 2001). Therefore, there have been many efforts actively ongoing to identify an alternative and complimentary medicine regulating specific target molecules associated with IBD.
Upon stimulation with TNF-α, disrupted epithelial cells in the intestinal mucosa perpetuate inflammatory process by secreting chemokines (Eckmann et al., 1993; Schuerer-Maly et al., 1994; Jung et al., 1995). Chemokines, during inflammation, attract and activate leukocytes at the site of inflammation and up-regulate adhesion molecules that are important for leukocyte trafficking. MCP-1 is considered to be one important chemokine regulating migration and infiltration of monocytes/macrophages. The important role of MCP-1 during intestinal inflammation has been demonstrated in recent studies in which markedly increased MCP-1 level is observed in the colon tissue of IBD patients (Reinecker et al., 1995). IL-8, a chemokine which attracts neutrophils, macrophages and T lymphocytes, has been also shown to be elevated in the mucosa of IBD patients (Mazzucchelli et al., 1994; Daig et al., 1996). The expression of TNF-α, MCP-1 and IL-8 that play an important role in the pathogenesis of IBD is dependent on NF-κB, a ubiquitous transcription factor involved in the regulation of proinflammatory gene expression (Manna and Aggarwal, 1998). Therefore, NF-κB may be a molecular target for drugs used in the treatment of IBD.
Mushrooms have been considered as an edible and medicinal resource for thousands of years. Grifola frondosa (GF) has been also used as a remedy for pain and inflammation in Southeast Asia (Mayell, 2001). Extensive studies have demonstrated that extract from fruiting body or liquid-cultured mycelium of GF exhibits considerable biological activities such as anti-tumor, anti-mutagenic, anti-hypertensive, anti-diabetic, hypolipidemic, and collagen biosynthesis-enhancing activities (Kubo et al., 1994; Mizuno and Zhuang, 1995; Shigesue et al., 2000; Mayell, 2001; Lee et al., 2003; Shomori et al., 2009). Previously, our group has also demonstrated that GF water extract (GFW) protected against carbon tetrachloride (CCl4)-induced liver injury (Lee et al., 2008a) and VEGF-induced ROS and ERK phosphorylation (Lee et al., 2008b).
In the present study, we examined the inhibitory effects and mechanism of action of GFW on intestinal inflammation by using in vitro bioassay model of IBD in which HT-29 cells were treated with TNF-α, and in vivo animal model of IBD, TNBS-induced colitis in rats.
Results
GFW inhibits TNBS-induced rat colitis
The rats treated with TNBS developed significant signs of colitis, bloody diarrhea and wasting conditions with sluggish and weak movement. In addition, TNBS induced stagnated body weight of rats in contrast to the weight gain in vehicle-treated control groups (Figure 1A). The weight of colon tissue per cm3 (between 5 and 6 cm proximal to the rectum) was increased by TNBS (Figure 1B). However, administration of the rats with GFW (orally, 1 g/kg) or 5-ASA (i.p. 100 mg/kg) significantly reversed the decrease of body weight and increase of colon weight associated with TNBS-induced colitis. Moreover, GFW significantly reduced colonic myeloperoxidase activity, which serves as a marker for tissue infiltration by neutrophils (Figure 1C).

GFW improves the clinical and morphological features of TNBS-induced colitis in rats. Colitis was induced by rectal administration of TNBS. The control group received 50% ethanol as a vehicle. A, the body weight was recorded daily from day 1 to day 6. The wet weight of colon (between 5 and 6 cm proximal to the rectum) was measured (B) and the myeloperoxidase activity of colon tissue (C) was measured by using the myeloperoxidase assay kit. The data represent the mean ± SEM for 5 rats per group. *P < 0.01 compared with vehicle-treated control group. #P < 0.01 compared with the TNBS-treated group.
GFW suppresses TNBS-induced TNF-α expression in rat colon
In histomorphometrical examinations, TNBS induced a significant ablation of mucosa, increased thickness of submucosa and total colonic walls. However, such histopathological changes were dramatically suppressed by the treatment with GFW. We also examined that GFW inhibits colonic TNF-α expression in TNBS-induced colitis model. In TNBS-treated rat colon, there was a marked increase of TNF-α-immunoreactive cells (over 10% of immunoreactivity) compared to untreated control group (Figure 2). However, the TNBS-induced TNF-α expression was dramatically inhibited in the colon tissue from GFW-treated group, of which effect was similar to that from 5-ASA-treated group.

GFW suppresses TNBS-induced TNF-α expression in rat colon. The colon tissues were either counterstained with hematoxylin and eosin (left column) or immunostained with TNF-α antibody (middle and right columns). A, B, and C, untreated control; D, E, and F, treated with TNBS; G, H, and I, treated with TNBS and 5-ASA; J, K, and L, treated with TNBS and GFW. The TNF-α -immunoreactive cells were presented as dark blue color. MM, muscularis mucosa; SL, submucosa layer; ML, muscle layer; TA, tunica adventitia. Scale bars = 160 µm.
GFW inhibits LPS-induced TNF-α secretion in TPA-differentiated U937 cells
TNF-α is a potent inducer of NF-κB transactivation, and it also regulates a variety of NF-κB-dependent gene expression including TNF-α itself. Since GFW showed an inhibitory activity against NF-κB activation, we further investigated the effects of GFW on NF-κB-dependent TNF-α production in response to bacterial LPS stimulation. As shown in Figure 3, in the TPA-differentiated U937 cells, LPS significantly increased TNF-α protein secretion. The co-treatment with GFW, however, significantly suppressed the TNF-α secretion.

Inhibitory effects of GFW on LPS-induced TNF-α secretion in the TPA-differentiated U937 cells. U937 cells were differentiated by the incubation with TPA (20 ng/ml) for 48 h, and then, stimulated with LPS for 18 h. Secreted TNF-α protein level in the culture supernatant was measured using a TNF-α ELISA kit. Data are expressed as the mean ± SEM of three independent experiments with duplicate. *P < 0.01 compared to untreated control group. #P < 0.01 compared to LPS-treated group.
Inhibitory effects of GFW on TNF-α-induced intestinal inflammation in HT-29 cells
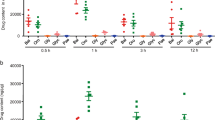
TNF-α-induced adhesion of monocytes to colon epithelial cells represents an in vitro model of intestinal inflammation (Thapa et al., 2009). As shown in Figures 4A and 4B, TNF-α significantly enhanced the adhesion of fluorescence-labeled U937 premonocytic cells to HT-29 cells. However, treatment with GFW (10, 50, 100 µg/ml) significantly inhibited the adhesion in a concentration-dependent manner.

Inhibitory effects of GFW on TNF-α-induced monocyte adhesion to HT-29 cells. HT-29 cells were pretreated with GFW or 5-ASA for 1 h prior to the treatment with 10 ng/ml TNF-α for an additional 3 h. Then, HT-29 cells were co-incubated with BCECF/AM-prelabeled U937 cells for 1 h at 37℃. A set of cells was examined under inverted fluorescence microscopy (A), whereas other cells were lysed in 0.1% Triton X-100 in 0.1 M Tris, and the fluorescence was measured by using a fluorometer (Fluostar optima) with excitation and emission at 485 nm and 520 nm, respectively (B). The bar graphs represent the mean ± SEM of four independent experiments. *P < 0.01 compared to untreated control group. #P < 0.01 compared to TNF-α-treated group.
Since increased level of chemokines, MCP-1 and IL-8, control monocyte recruitment to colon epithelial cells during inflammation (Kolios et al., 1999), we examined the effects of GFW on TNF-α-induced MCP-1 and IL-8 mRNA level. As shown in Figure 5, treatment of HT-29 cells with TNF-α significantly increased MCP-1 mRNA (Figures 5A and 5B) and protein (Figure 5C) levels, which was significantly suppressed by the treatment with GFW. In addition, the mRNA expression of IL-8, an important cytokine related to the pathogenesis of IBD was increased by TNF-α, which was significantly suppressed by GFW in a concentration-dependent manner (Figures 5A and 5B). The inhibitory effects of GFW at 100 µg/ml concentration on the adhesion and chmokine expression were similar to that of 20 mM (3.06 mg/ml) of 5-ASA, a commonly used drug for IBD.

Inhibitory effect of GFW on TNF-α-induced MCP-1 and IL-8 expression in HT-29 cells. Serum-starved HT-29 cells were pretreated with GFW or 5-ASA for 1 h prior to the treatment with 10 ng/ml TNF-α. The MCP-1 and IL-8 mRNA expression level was quantified by RT-PCR (A). The bar graph in (B) represents the relative density of MCP-1 and IL-8 mRNA. In the experiment of (C), culture supernatant of drug-treated HT-29 cells was measured for the MCP-1 protein secretion using an MCP-1 ELISA kit according to the manufacturer's protocol. Data are expressed as the mean ± SEM of four independent experiments. *P < 0.01 compared to untreated control group. #P < 0.01 compared to TNF-α-treated group.
GFW inhibits TNF-α-induced ROS generation and NF-κB activation in HT-29 cells
Oxygen free radicals are clearly involved in the pathogenesis of various inflammatory diseases including IBD (Grisham and Granger, 1988). Since TNF-α is known to induce a rapid increase of intracellular ROS levels (Matthews et al., 1987), we investigated whether GFW suppresses the TNF-α-induced ROS production by using cytochrome c reduction assay. As shown in Figure 6A, GFW significantly suppressed the TNF-α (10 ng/ml)-induced ROS production in HT-29 cells. In a DPPH assay, radical scavenging activity of GFW was strong and concentration-dependent (Figure 6B).

The effects of GFW on TNF-α-induced ROS generation in HT-29 cells (A) and DPPH-generated free radical (B). In the experiment of (A), the cells were treated with GFW for 1 h prior to TNF-α (10 ng/ml) stimulation. After 3 h, cytochrome c (80 µM) was added to each well and incubated for 15 min at 37℃. The level of cytochrome c reduction represents cellular ROS production. The absorbance was read at 550 nm by a spectrophotometer. Data are expressed as the mean ± SEM of three independent experiments. In the experiment of (B), GFW was incubated with DPPH for 30 min, the absorbance at 517 nm due to DPPH radical was determined. DPPH radical scavenging activity was calculated from the following equation in which H and Ho were optical density of solvent with and without sample, respectively. Radical scavenging activity (%) = {(Ho - H)/Ho} × 100. The data represent the mean ± SEM. *P < 0.01 compared to untreated control group. #P < 0.01 compared to TNF-α-treated group.
Since it has been well known that intracellular ROS leads to activation of the transcription factor, NF-κB, which is a major regulator of inflammatory gene expression (Wajant et al., 2003), we also examined the inhibitory effect of GFW on NF-κB activity. In HT-29 cells transfected with NF-κB-Luc plasmid, TNF-α (10 ng/ml) induced a significant increase (14 fold) in NF-κB transcriptional activity (Figure 7). However, GFW concentration-dependently inhibited the TNF-α-induced NF-κB transcriptional activity. Such effect of GFW at 100 µg/ml concentration was stronger than that of 20 mM 5-ASA.

Inhibitory effect of GFW on TNF-α-induced NF-κB luciferase activity in HT-29 cells. The HT-29 cells that were transfected with NF-κB reporter gene were treated with GFW or 5-ASA for 1 h followed by 3 h treatment with TNF-α. Cells were then lysed, and NF-κB gene transcription activity was measured by dual-luciferase assay kit according to the manufacturer's protocol. Data are means ± SEM from four independent experiments. *P < 0.01 compared to untreated control and #P < 0.01 compared to TNF-α-treated group.
Phenolic component in GFW
In our previous study, we reported that GFW contains high amounts of phenolic compounds, and it has been reported that less than 28 phenolic compounds were detected in edible and medicinal mushrooms (Kim et al., 2008b). In this study, in order to identify the types of phenolic compounds in GFW, we performed HPLC using 32 phenolic compound standards. As shown in Table 1, GFW also contained many different types of phenolic compounds including pyrogallol, caffeic acid, myricetin, protocatechuic acid, veratric acid, naringin, and kaempferol.
Discussion
Prolonged or inadequate activation of the intestinal immune system and increased production of proinflammatory cytokines in the intestinal mucosa are thought to be a pivotal factor in the pathogenesis of intestinal inflammation (Schreiber et al., 1992; Sartor, 1997). Although many drugs have been used in the treatment of IBD, none has, so far, been shown to modify the natural history of the diseases or to maintain a stable remission over time (Hanauer and Sandborn, 2001). Recently, it has been reviewed that alternative and complementary medicine improves inflammatory symptoms, and thus, quality of life by preventing the inflammatory cascade (Clarke and Mullin, 2008).
Oxidant-mediated injury plays an important role in the pathophysiology of IBD. TNF-α, a pro-inflammatory cytokine, is known to induce a rapid increase of intracellular ROS levels (Matthews et al., 1987), and upregulate the expression of chemokines (Sawa et al., 2007). Our results clearly showed that GFW inhibited the TNF-α-induced ROS level and monocyte adhesion to epithelial cells. These results are consistent with previous findings that suppression of ROS inhibits leukocyte-epithelial cell adhesion during inflammatory process (Kim et al., 2008a). Furthermore, our results indicate that GFW contains strong antioxidant components that inhibit ROS production by TNF-α, and thus, ultimately suppress the TNF-α-induced recruitment of leukocytes to epithelial cells. Such innate immune modulating activity of Grifola frondosa fruit body has been demonstrated in its D-fraction which contains high molecular weight polysaccharides (Lee et al., 2003). The D fraction which is composed of (1→3)-branched (1→6)-β-glucan is shown to activate natural killer cells via macrophage-derived IL-12, and T cells (Kodama et al., 2002). In addition, D fraction also stimulates special population of macrophages in a way that it stimulates the production of TNF-α only in splenic macrophages, not in peritoneal macrophages (Shigesue et al., 2000).
In addition to the polysaccharides, we have previously reported that GFW contains high amounts of polyphenols (Lee et al., 2008b) that have been shown to be potentially immunomodulating agents (Shapiro et al., 2007). The dietary polyphenols such as resveratrol, epigallocatechin, curcumin, and boswellia have been demonstrated to prevent and treat animal models of IBD (Clarke and Mullin, 2008). Likewise, our results also suggest that mushroom polyphenols have ability to prevent the intestinal inflammation. Recently, pyrogallol, the major polyphenol in GFW, has been reported to regulate leukocyte spreading behavior (Kori et al., 2009) as well as expression of proinflammatory genes in bronchial epithelial cells (Nicolis et al., 2008). Not only pyrogallol but also caffeic acid (Prasad et al., 2009), myricetin (Pandey et al., 2009), and naringin (Rajadurai and Prince, 2009) are well known to prevent oxidative stress.
NF-κB is one of the redox-dependent transcription factors and plays a critical role in the regulation of the expression and function of various genes involved in inflammation. Activation of NF-κB has been demonstrated in situ in macrophages and intestinal epithelial cells in the inflamed mucosa from IBD patients (Rogler et al., 1998) and animal models (Sanchez-Hidalge et al., 2007). The expression of MCP-1 which is upregulated in the colon of IBD patients is dependent on the activation of NF-κB. Our results showed that GFW suppressed NF-κB transcriptional activity as well as MCP-1 expression. Similarly, the expression of IL-8, one of the CXC chemokines attracting neutrophils as well as macrophages and T lymphocytes, has been suggested to be NF-κB-dependent. The TNF-α-induced IL-8 increase was also suppressed by GFW. Furthermore, TNF-α is an inducer of NF-κB activation, and it is also a NF-κB-dependent gene product (Manna and Ramesh, 2005). As expected from the result of NF-κB suppression, GFW significantly blocked LPS-induced TNF-α secretion. The suppression of NF-κB activation as well as down-regulated MCP-1 and IL-8 by GFW was corresponded to the inhibition of monocyte adhesion to epithelial cells. The results suggest that the action of polyphenol-containing GFW in blocking the migration and adhesion of monocytes to the colon epithelial cells are mediated through regulation of NF-κB activity. These results are consistent with previous findings of the inhibitory effects of polyphenols on NF-κB activity associated with inflammation (Kundu and Surh, 2004; Ukil et al., 2006).
Such effects of GFW on in vitro model of IBD was confirmed in an in vivo animal model of IBD, TNBS-induced rat colitis, which is a well established model of intestinal inflammation with some resemblance to human IBD. The histopathological lesions of the colon by TNBS which are generally documented as ulceration, mucus cell depletion, inflammatory cell infiltration, and edematous changes were suppressed by oral administration of GFW (1 g/kg). Also, TNBS-increased myeloperoxidase activity, a quantitative index of inflammation in a damaged tissue (Cetinkaya et al., 2005, 2006), was inhibited by GFW. This result corresponds to previous studies that antioxidants can suppress intestinal inflammation (Sekizuka et al., 1988; Cetinkaya et al., 2005, 2006; Necefli et al., 2006). Furthermore, our study showed that the TNBS-induced TNF-α expression in rat colon was significantly suppressed by GFW. These results suggest that GFW prevents production of proinflammatory cytokine TNF-α as well as the action of TNF-α in the colon.
In in vitro studies, our present study showed that the inhibitory effects of GFW were even better than the case of 5-ASA, a well known anti-inflammatory drug for IBD. Furthermore, in the TNBS-induced rat colitis, the inhibitory effects of orally administered GFW were similar to those of 5-ASA (i.p.). Taken together, the results strongly suggest that GFW is a valuable medicinal food for IBD treatment, and thus may be used as an alternative medicine for IBD.
Methods
Preparation of GFW
Dried GF fruit body purchased from Chiba market (Tokyo, Japan) was extracted with twenty-fold water for 24 h at 40℃. The extract was filtered with Whatman filter paper No. 2. The filtrate was concentrated by a vacuum evaporator, freeze dried, and preserved at -20℃ until use. About 49.2 g of dried water extract was obtained from 100 g dried GF fruit body. The extract was re-suspended in distilled water, and used for experiments.
TNBS-induced experimental rat colitis
Sprague-Dawley rats (7-8 weeks old) were purchased from Samtaco Bio Korea (Osan, Korea). Animal experiments were performed according to the institutional guidelines of the Institute of Laboratory Animal Resources (1996) and Yeungnam University for the care and use of laboratory animals.
Rats were fasted (able to drink ad libitum) for 24 h before induction of colitis. They were then anesthetized lightly using diethyl ether. Using a polyethylene catheter fitted onto a 1-ml syringe, rats received injections slowly with 0.8 ml of 5% TNBS in 50% (v/v) ethanol into the lumen of the colon (8 cm proximal to the anus through the rectum), and they were kept in a vertical position for 60 s before being returned to their cages. Rats in the control group were handled similarly, but 50% (v/v) ethanol alone was administrated instead. To investigate the effect of the drugs, rats were orally administrated with GFW 1 g/kg/day for 5 days after the TNBS administration. In our experiments, 5-ASA, an active metabolite of sulfasalazine that is a commonly used drug for IBD, was administered intraperitoneally. All rats were sacrificed on day 5 after the TNBS administration. The macroscopic ulceration and severity of colitis was evaluated by two independent investigators who were blinded to the treatment. The colon tissues from 5 to 7 cm proximal to the rectum were cut out and used for myeloperoxidase activity assay and histological examinations.
Myeloperoxidase activity assay
Myeloperoxidase serves as a marker for tissue neutrophil infiltration. To measure myeloperoxidase activity, we used a myeloperoxidase assay kit (HK210; Hycult biotechnology, Netherlands). The 1-cm segment of dissected colon tissues were washed in cold PBS (pH 7.4), weighed, added 200 µl of ice-cold lysis buffer (pH7.4, 200 mM NaCl, 5 mM EDTA, 10 mM Tris, 10% glycerol, 1 mM PMSF, 1 µg/ml leupeptide, 28 µg/ml aprotinine) to 10 mg of tissue, and homogenized for 30 s using a tissue homogenizer at 4℃ (Biospec Products Inc., Bartlesville, Oklahoma). The homogenate was centrifuged at 1500 g at 4℃ for 15 min and the supernatant was measured by using an ELISA kit (HK210) according to the supplier's protocol. Quantification was performed by determination of the optical density within 30 min, using a microplate reader set to 450 nm.
Immunohistochemistry
All trimmed colons were fixed in 10% neutral buffered formalin. After paraffin embedding, 3-4 µm sections were prepared. Representative sections were stained with hematoxylin and eosin (H&E) for light microscopical examination. After de-paraffinization and citrate buffer antigen (epitope) retrieval pretreatment (Norton et al., 1994; Han et al., 2005), sections were incubated with methanol and 0.3% H2O2 for 30 min for blocking endogenous peroxidase activity at room temperature. After rinse in 0.01 M PBS (pH 7.2) for 3 times, the sections were incubated with normal horse serum blocking solution (Vector Lab. Inc., CA. Dilution 1:100) for 1 h in a humidity chamber to block non-specific binding of immunoglobulin at room temperature. Then the sections were rinsed and incubated with primary TNF-α antiserum (Santa Cruz Biotechnology, CA; Dilution, 1:200) for overnight at 4℃ in humidity chamber. Then the sections were incubated with biotinylated universal secondary antibody (Vector Lab. Inc., CA. Dilution 1:50) for 1 h at room temperature in humidity chamber. Then, the sections were serially incubated with avidin-biotin complex reagents (Vectastain Elite ABC Kit, Vector Lab. Inc., CA. Dilution 1:50) for 1 h, rinsed in 0.01M PBS for 3 times and incubated in peroxidase substrate kit (Vector Lab. Inc., CA) for 5 min at room temperature. The sections were counterstained with Mayer's hematoxylin solution, dehydrated through 95% ethanol for 2 min, 100% ethanol for 3 times, and cleared in xylene for two times, and covered with permanent mounting medium and observed under light microscope (Nikon, Japan).
Cell culture
HT-29 cells were grown in RPMI 1640 medium supplemented with 10% FBS and 200 IU/ml penicillin in a humidified incubator under 5% CO2/95% air. The culture medium was replaced every other day. After reaching at confluency, the cells were subcultured by using trysinization with trypsin-EDTA solution. U937 human pre-monocytic cells were maintained in a RPMI 1640 medium supplemented with 10% FBS, 1 mM sodium pyruvate, 100 IU/ml penicillin and 100 µg/ml of streptomycin. For differentiation of U937 cells to monocytes, the cells were pretreated with 20 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA) for 48 h.
ELISA
MCP-1 and TNF-α levels in HT-29 and U937 culture supernatant, respectively, was measured by using an ELISA kit specific to MCP-1 and TNF-α (R&D Systems) according to the supplier's protocol. Quantification was performed by determination of the optical density within 30 min, using a microplate reader set to 450 nm. TPA-differentiated U937 cells were pretreated with GFW or 5-ASA for 24 h prior to incubation with 100 ng/ml LPS for an additional 18 h.
Monocyte-epithelial cell adhesion assay
Monocyte to colonic epithelial cell adhesion was evaluated using the human U937 human pre-monocytic cells (Kobayashi et al., 2006), which were prelabeled with 2',7'-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF/AM, 10 µg/ml) for 1 h at 37℃. HT-29 cells cultured in a 24-well plate were pretreated with GFW or 5-aminosalicylic acid (5-ASA) for 1 h prior to being incubated with TNF-α (10 ng/ml) for an additional 3 h. Cells were then co-incubated with BCECF/AM-prelabeled U937 cells (1 × 106 cells/ well) for 30 min at 37℃. Non-adhering U937 cells were removed, and the HT-29 cells were washed twice with PBS. A set of cells were examined under an inverted fluorescence microscope equipped with a digital camera (TE2000-U, Nikon, Japan). Others were lysed in 0.1% Triton X-100 in Tris (0.1 M), and the fluorescence was measured using a fluorescence-detecting microplate reader (Fluostar Optima, BMG LABTECH GmbH, Germany) at excitation and emission wavelengths of at 485 and 520 nm, respectively.
RT-PCR
Serum-starved HT-29 cells were pretreated with GFW or 5-ASA for 1 h prior to the incubation with 10 ng/ml TNF-α for an additional 3 h. Total cellular RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA) and RT-PCR was performed as previously described (Beak et al., 2004). Briefly, cDNA was synthesized using a Ready-To-Go T-Primed First Strand kit (Amersham Biosciences), and PCR was performed in the presence of 0.5 U Taq DNA polymerase (Takara, Japan) using primers for human MCP-1, IL-8, and GAPDH. Amplification conditions were; denaturation at 94℃ for 4 min for the first cycle and for 1 min for the second cycle. The annealing and extension conditions used were: MCP-1 at 58℃ for 45 s and 35 amplification cycles of 72℃ for 60 s, IL-8 at 65℃ for 45 s and 19 amplification cycles of 72℃ for 90 s, and GAPDH 58℃ for 45 s and 30 amplification cycles of 72℃ for 45 s. Final extensions were performed at 72℃ for 10 min. The sequences of the primers used were as follows: human MCP-1 (sense 5'-ATGAAAGTCTCTGCCGCCCTT-3' antisense 5-TCAAGTCTTCGGAGTTTGGGT-3'); IL-8 (sense 5'-ATGACTTCCAAGCTGGCCGTGGCT-3' antisense 5'-TCTCAGCCCTCTTCAAAAACTTCTC-3'); GAPDH (sense 5'-GGTGAAGGTCGGAGTCAACG-3', antisense: 5'-CAAAGTTGTCATGGATGACC-3'). PCR products so obtained were separated on 2% agarose gels containing ethidium bromide (0.5 µg/ml), and visualized and photographed using a gel documentation system (UVP, Cambridge, UK). The mRNA levels of all genes were normalized versus GAPDH. The lengths of amplicons were 300, 289, and 496 base pairs for MCP-1, IL-8, and GAPDH, respectively.
Cytochrome c reduction assay
The level of cytochrome c reduction was measured to assess cellular reactive oxygen species (ROS) production (Ding et al., 2007). HT-29 cells (1 × 105 cells/well) cultured in 96-well plate were pre-treated with GFW for 1 h, and then, incubated with TNF-α (10 ng/ml) for an additional 3 h. Cells were then washed with PBS, treated with 80 µM of cytochrome c in PBS for 15 min at 37℃, and then cooled to 0℃. The absorbance was read at 550 nm using a spectrophotometer (UV-1601, Shimadzu, Japan). The amount of superoxide anion released was determined on the basis of cytochrome c reduction and results are expressed as nmol/well/min.
Measurement of 2,2-diphenyl-1-picrylhydrazyl radical-scavenging activity
Radical-scavenging activity was measured by means of a 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay (Shirwaikar et al., 2006). GFW was dissolved and diluted in distilled water at various concentrations. DPPH in ethanol (200 µM) was mixed with GFW for 30 min at room temperature. The absorbance of the resulting solution at 517 nm was measured by using a spectrophotometer.
NF-κB reporter gene dual-luciferase assay
HT-29 cells were transfected with 1 µg/ml of the NF-κB luciferase construct (firefly luciferase) in conjunction with 0.2 µg/ml of the pRL-TK (renilla luciferase) as a transfection control using GeneJammer transfection reagent (Staratagene, CA) according to the manufacturer's instructions. Cells were incubated with transfection mixture at 37℃ for 3 h, mixed with the same volume of RPMI 1640 growth medium, and kept in an incubator at 37℃ overnight. These cells were then plated onto 24-well plate. After 24 h, the cells were pretreated with GFW or 5-ASA for 1 h, and then, incubated with 10 ng/ml TNF-α for an additional 3 h. After indicated time, the cells were washed with PBS and then lysed by repeated freezing and thawing. Cells were then scraped gently, and the lysates were centrifuged at 10,000 rpm for 5 min. Firefly and renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay Kit (Promega Corporation, Madison, WI) on a Turner TD20/20 luminometer (Turner Biosystems, CA).
Polyphenolic compound measurement
The measurement of phenolic compounds in GFW was performed by HPLC with standard curves of 32 phenolic compounds, gallic acid, 5-sulfosalicylic acid, pyrogallol, homogentisic acid, protocatechuic acid, gentisic acid, chlorogenic acid, p-hydroxybenzoic acid, (+)catechin, vanillic acid, syringic acid, caffeic acid, vanillin, p-coumaric acid, rutin, ferulic acid, m-coumaric acid, salicylic acid, hesperidin, o-coumaric acid, myricetin, resveratrol, quercetin, t-cinnamic acid, naringenin, hesperetin, formononetin, biochanin A, β-resorcylic acid, naringin, kaempferol, and veratric acid. GFW was dissolved in 10 ml of 80% aqueous methanol (HPLC grade), filtered through 0.45 µm nylon membrane filter. The filtrate (20 µl) was loaded on the HPLC (Shimadzu SPD-M10A) system with a photodiode array detector (Tokyo, Japan) equppied with a Midas autoinjector. The analysis of each polyphenol concentration was performed according to the method previously reported (Kim et al., 2008b).
Statistical analysis
The data are expressed as means ± SEM of three independent experiments and analyzed using one-way analysis of variance (ANOVA) and the Student's t-test. P values of < 0.05 were considered statistically significant.
Abbreviations
- 5-ASA:
-
5-aminosalicylic acid
- BCECF/AM:
-
2',7'-bis (2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester
- DPPH:
-
2,2-diphenyl-1-picrylhydrazyl
- GFW:
-
water extract of Grifola frondosa
- IBD:
-
inflammatory bowel disease
- MCP-1:
-
monocyte chemoattractant protein-1
- ROS:
-
reactive oxygen species
- TNBS:
-
trinitrobenzene sulfonic acid
- TPA:
-
12-O-tetradecanoylphorbol-13-acetate
References
Autschbach F, Schurmann G, Qiao L, Merz H, Wallich R, Meuer SC . Cytokine messenger RNA expression and proliferation status of intestinal mononuclear cells in noninflamed gut and Crohn's disease . Virchows Arch 1995 ; 426 : 51 - 60
Babu D, Lee JS, Park SY, Thapa D, Choi MK, Kim AR, Park YJ, Kim JA . Involvement of NF-κB in the inhibitory actions of Platycarya strobilacea on the TNF-α-induced monocyte adhesion to colon epithelial cells and chemokine expression . Arch Pharm Res 2008 ; 31 : 727 - 735
Beak SM, Lee YS, Kim JA . NADPH oxidase and cyclooxygenase mediate the ultraviolet B-induced generation of reactive oxygen species and activation of nuclear factor-kappaB in HaCaT human keratinocytes . Biochimie 2004 ; 86 : 425 - 429
Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT . Tumour necrosis factor alpha in stool as a marker of intestinal inflammation . Lancet 1992 ; 339 : 89 - 91
Cetinkaya A, Bulbuloglu E, Kantarceken B, Ciralik H, Kurutas EB, Buyukbese MA, Gumusalan Y . Effects of Lcarnitine on oxidant/antioxidant status in acetic acid-induced colitis . Dig Dis Sci 2006 ; 51 : 488 - 494
Cetinkaya A, Bulbuloglu E, Kurutas EB, Ciralik H, Kantarceken B, Buyukbese MA . Beneficial effects of N-acetylcysteine on acetic acid-induced colitis in rats . Tohoku J Exp Med 2005 ; 206 : 131 - 139
Clarke JO, Mullin GE . A review of complementary and alternative approaches to immunomodulation . Nutr Clin Pract 2008 ; 23 : 49 - 62
Daig R, Andus T, Aschenbrenner E, Falk W, Scholmerich J, Gross V . Increased IL-8 expression in the colon mucosa of patients with inflammatory bowel disease . Gut 1996 ; 38 : 216 - 222
Ding SZ, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden-Kramer B . Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells . Infect Immun 2007 ; 75 : 4030 - 4039
Eckmann L, Jung HC, Schürer-Maly C, Panja A, Morzycka-Wroblewska E, Kagnoff MF . Differential cytokine expression by human intestinal epithelial cell lines: regulated expression of interleukin 8 . Gastroenterology 1993 ; 105 : 1689 - 1697
Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT . Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota . Immunol Rev 2005 ; 206 : 260 - 276
Grisham MB, Granger DN . Neutrophil-mediated mucosal injury. Role of reactive oxygen metabolites . Dig Dis Sci 1988 ; 33 : 6S - 15S
Hanauer SB, Sandborn W . Management of Crohn's disease in adults . Am J Gastroenterol 2001 ; 96 : 635 - 643
Han SR, Son SK, Yang YS, Kim WS . TNFα and TNFR2 immunohistochemistry during ovarian follicular development and atresia in the rat . Korean J Anat 2005 ; 38 : 553 - 559
Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF . A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion . J Clin Invest 1995 ; 95 : 55 - 65
Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM . Tuberculosis associated with infliximab, a tumor necrosis factor-neutralizing agent . N Engl J Med 2001 ; 345 : 1098 - 1104
Kim H, Hwang JS, Woo CH, Kim EY, Kim TH, Cho KJ, Kim JH, Seo JM, Lee SS . TNF-alpha-induced up-regulation ofintercellular adhesion molecule-1 is regulated by a Rac-ROS-dependent cascade in human airway epithelial cells . Exp Mol Med 2008a ; 40 : 167 - 175
Kim MY, Seguin P, Ahn JK, Kim JJ, Chun SC, Kim EH, Seo SH, Kang EY, Kim SL, Park YJ, Ro HM, Chung IM . Phenolic compound concentration and antioxidant activities of edible and medicinal mushrooms from Korea . J Agric Food Chem 2008b ; 56 : 7265 - 7270
Kim YS, Morgan MJ, Choksi S, Liu ZG . TNF induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death . Mol Cell 2007 ; 6 : 675 - 687
Kobayashi N, Ikesue A, Majumdar S, Siahaan TJ . Inhibition of e-cadherin-mediated homotypic adhesion of Caco-2 cells: a novel evaluation assay for peptide activities in modulating cell-cell adhesion . J Pharmacol Exp Ther 2006 ; 317 : 309 - 316
Kodama N, Komuta K, Sakai N, Nanba H . Effects of D-Fraction, a polysaccharide from Grifola frondosa on tumor growth involve activation of NK cells . Biol Pharm Bull 2002 ; 25 : 1647 - 1650
Kolios G, Wright KL, Jordan NJ, Leithead JB, Robertson DA, Westwick J . C-X-C and C-C chemokine expression and secretion by the human colonic epithelial cell line, HT-29: differential effect of T lymphocyte-derived cytokines . Eur J Immunol 1999 ; 29 : 530 - 536
Komatsu M, Kobayashi D, Saito K, Furuya D, Yagihashi A, Araake H, Tsuji N, Sakamaki S, Niitsu Y, Watanabe N . Tumor necrosis factor-alpha in serum of patients with inflammatorybowel disease as measured by a highly sensitive immuno-PCR . Clin Chem 2001 ; 47 : 1297 - 1301
Kori S, Namiki H, Suzuki K . Biphasic regulation of polymorphonuclear leukocyte spreading by polyphenolic compounds with pyrogallol moieties . Int Immunopharmacol 2009 ; [Epub ahead of print]
Kubo K, Aoki H, Nanba H . Anti-diabetic activity present in the fruit body of Grifola frondosa (maitake) . Biol Pharm Bull 1994 ; 17 : 1106 - 1110
Kundu JK, Surh YJ . Molecular basis of chemoprevention by resveratrol: NF-kappaB and AP-1 as potential targets . Mutat Res 2004 ; 555 : 65 - 80
Lee BC, Bae JT, Pyo HB, Choe TB, Kim SW, Hwang HJ, Yun JW . Biological activities of the polysaccharides produced from submerged culture of the edible Basidiomycete Grifola frondosa . Enzyme Microb Technol 2003 ; 32 : 574 - 581
Lee JS, Kim HS, Lee YJ, Yong CH, Choi HG, Han GD, Kim JA, Lee JS . Hepatoprotective effect of Grifola frondosa water extract on carbon tetrachloride-induced liver injury in rats . Food Sci Biotechnol 2008a ; 17 : 203 - 207
Lee JS, Park BC, Ko YJ, Choi MK, Choi HG, Yong CS, Lee JS, Kim JA . Grifola frondosa (Maitake) water extract inhibits VEGF-induced angiogenesis through inhibition of reactive oxygen species and ERK phosphorylation . J Med Food 2008b ; 11 : 643 - 651
Manna SK, Aggarwal BB . IL-13 suppresses TNF-induced activation of nuclear factor-kappa B, activation protein-1, and apoptosis . J Immunol 1998 ; 61 : 2863 - 2872
Manna SK, Ramesh GT . Interleukin-8 induces nuclear transcription factor-kappaB through a TRAF6-dependent pathway . J Biol Chem 2005 ; 280 : 7010 - 7021
Matthews N, Neale ML, Jackson SK, Stark JM . Tumour cell killing by tumour necrosis factor: inhibition by anaerobic conditions, free-radical scavengers and inhibitors of arachidonate metabolism . Immunology 1987 ; 62 : 153 - 155
Mazzucchelli L, Hauser C, Zgraggen K, Wagner H, Hess M, Laissue JA, Mueller C . Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation . Am J Pathol 1994 ; 144 : 997 - 1007
Mayell M . Maitake extracts and their therapeutic potential . Altern Med Rev 2001 ; 6 : 48 - 60
Mizuno T, Zhuang C . Maitake, Grifola frondosa: pharmacological effects . Food Rev Intl 1995 ; 11 : 135 - 149
Murch SH, Braegger CP, Walker-Smith JA, MacDonald TT . Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease . Gut 1993 ; 34 : 1705 - 1709
Necefli A, Tulumoglu B, Giris M, Barbaros U, Gündüz M, Olgaç V, Güloğlu R, Toker G . The effect of melatonin on TNBS-induced colitis . Dig Dis Sci 2006 ; 51 : 1538 - 1545
Nicolis E, Lampronti I, Dechecchi MC, Borgatti M, Tamanini A, Bianchi N, Bezzerri V, Mancini I, Giri MG, Rizzotti P, Gambari R, Cabrini G . Pyrogallol, an active compound from the medicinal plant Emblica officinalis, regulates expression of pro-inflammatory genes in bronchial epithelial cells . Int Immunopharmacol 2008 ; 8 : 1672 - 1680
Norton AJ, Jordan S, Yeomans P . Brief, high-temperature heat denaturation (pressure cooking): a simple and effective method of antigen retrieval for routinely processed tissues . J Pathol 1994 ; 173 : 371 - 379
Pandey KB, Mishra N, Rizvi SI . Protective role of myricetin on markers of oxidative stress in human erythrocytes subjected to oxidative stress . Nat Prod Commun 2009 ; 4 : 221 - 226
Podolsky DK . Inflammatory bowel disease . N Engl J Med 2002 ; 347 : 417 - 429
Prasad NR, Jeyanthimala K, Ramachandran S . Caffeic acid modulates ultraviolet radiation-B induced oxidative damage in human blood lymphocytes . J Photochem Photobiol B 2009 ; 95 : 196 - 203
Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand RA, Podolsky DK, Sands BE, Braakman T, DeWoody KL, Schaible TF, van Deventer SJ . Infliximab for the treatment of fistulas in patients with Crohn's disease . N Engl J Med 1999 ; 340 : 1398 - 1405
Rajadurai M, Prince PS . Naringin ameliorates mitochondrial lipid peroxides, antioxidants and lipids in isoproterenol-induced myocardial infarction in wistar rats . Phytother Res 2009 ; 23 : 358 - 362
Reimund JM, Wittersheim C, Dumont S, Muller CD, Kenney JS, Baumann R, Poindron P, Duclos B . Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn's disease . Gut 1996 ; 39 : 684 - 689
Reinecker HC, Loh EY, Ringler DJ, Mehta A, Rombeau JL, Mac Dermott RP . Monocyte-chemoattractant protein 1 gene expression in intestinal epithelial cells and inflammatory bowel disease mucosa . Gastroenterology 1995 ; 108 : 40 - 50
Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Schölmerich J, Gross V . Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa . Gastroenterology 1998 ; 115 : 357 - 369
Sartor RB . Pathogenesis and immune mechanisms of chronic inflammatory bowel disease . Am J Gastroenterol 1997 ; 92 : 58 - 118
Sánchez-Hidalgo M, Martín AR, Villegas I, de la Lastra CA . Rosiglitazone, a PPARgamma ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats . Eur J Pharmacol 2007 ; 562 : 247 - 258
Sawa Y, Sugimoto Y, Ueki T, Ishikawa H, Sato A, Nagato T, Yoshida S . Effects of TNF-alpha on leukocyte adhesion molecule expressions in cultured human lymphatic endothelium . J Histochem Cytochem 2007 ; 55 : 721 - 733
Schreiber S, Raedler A, Stenson WF, MacDermott RP . The immunology of inflammatory bowel disease . Gastroenterol Clin North Am 1992 ; 21 : 451 - 502
Schuerer-Maly CC, Eckmann L, Kagnoff MF, Falco MT, Maly FE . Colonic epithelial cell lines as a source of interleukin-8: stimulation by inflammatory cytokines and bacterial lipopolysaccharide . Immunology 1994 ; 81 : 85 - 91
Sekizuka E, Grisham MB, Li MA, Deitch EA, Granger DN . Inflammation-induced intestinal hyperemia in the rat: role of neutrophils . Gastroenterology 1988 ; 95 : 1528 - 1534
Shapiro H, Singer P, Halpern Z, Bruck R . Polyphenols in the treatment of inflammatory bowel disease and acute pancreatitis . Gut 2007 ; 56 : 426 - 435
Shigesue K, Kodama N, Nanba H . Effects of maitake (Grifola frondosa) polysaccharide on collagen-induced arthritis in mice . Jpn J Pharmacol 2000 ; 84 : 293 - 300
Shirwaikar A, Shirwaikar A, Rajendran K, Punitha IS . In vitro antioxidant studies on the benzyl tetra isoquinoline alkaloid berberine . Biol Pharm Bull 2006 ; 29 : 1906 - 1910
Shomori K, Yamamoto M, Arifuku I, Teramachi K, Ito H . Antitumor effects of a water-soluble extract from Maitake (Grifola frondosa) on human gastric cancer cell lines . Oncol Rep 2009 ; 22 : 615 - 620
Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ . A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease . N Engl J Med 1997 ; 337 : 1029 - 1035
Thapa D, Lee JS, Park MA, Cho MY, Park YJ, Choi HG, Jeong TC, Kim JA . Inhibitory effects of clotrimazole on TNF-alpha-induced adhesion molecule expression and angiogenesis . Arch Pharm Res 2009 ; 32 : 593 - 603
Ukil A, Maity S, Das PK . Protection from experimental colitis by theaflavin-3,3'-digallate correlates with inhibition of IKK and NF-kappaB activation . Br J Pharmacol 2006 ; 149 : 121 - 131
Wajant H, Pfizenmaier K, Scheurich P . Tumor necrosis factor signaling . Cell Death Differ 2003 ; 10 : 45 - 65
Acknowledgements
This work was supported by a grant No. RTI04-01-04 from the Regional Technology Innovation Program of the Ministry of Commerce, Industry, and Energy (MOCIE) and a grant No. R11-2007-040-02004-0 from Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MOST).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, J., Park, SY., Thapa, D. et al. Grifola frondosa water extract alleviates intestinal inflammation by suppressing TNF-α production and its signaling. Exp Mol Med 42, 143–154 (2010). https://doi.org/10.3858/emm.2010.42.2.016
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2010.42.2.016
Keywords
This article is cited by
-
Pulsatilla decoction suppresses matrix metalloproteinase-7-mediated leukocyte recruitment in dextran sulfate sodium-induced colitis mouse model
BMC Complementary Medicine and Therapies (2022)
-
Protective effects of Pogostemon cablin Bentham water extract on inflammatory cytokine expression in TNBS-induced colitis in rats
Archives of Pharmacal Research (2014)
-
Chinese Herbal Medicines in the Treatment of IBD and Colorectal Cancer: A Review
Current Treatment Options in Oncology (2014)
-
Lactobacillus plantarum Lipoteichoic Acid Alleviates TNF-α-Induced Inflammation in the HT-29 Intestinal Epithelial Cell Line
Molecules and Cells (2012)
-
Le Grifola frondosa (maïtaké) : un régulateur du syndrome métabolique: poids, cholestérol, glycémie et hypertension artérielle; accessoirement un immunostimulant
Phytothérapie (2011)
