
Abstract
Although a common cause of disease, copy number variants (CNVs) have not routinely been identified from next-generation sequencing (NGS) data in a clinical context. This study aimed to examine the sensitivity and specificity of a widely used software package, ExomeDepth, to identify CNVs from targeted NGS data sets. We benchmarked the accuracy of CNV detection using ExomeDepth v1.1.6 applied to targeted NGS data sets by comparison to CNV events detected through whole-genome sequencing for 25 individuals and determined the sensitivity and specificity of ExomeDepth applied to these targeted NGS data sets to be 100% and 99.8%, respectively. To define quality assurance metrics for CNV surveillance through ExomeDepth, we undertook simulation of single-exon (n=1000) and multiple-exon heterozygous deletion events (n=1749), determining a sensitivity of 97% (n=2749). We identified that the extent of sequencing coverage, the inter- and intra-sample variability in the depth of sequencing coverage and the composition of analysis regions are all important determinants of successful CNV surveillance through ExomeDepth. We then applied these quality assurance metrics during CNV surveillance for 140 individuals across 12 distinct clinical areas, encompassing over 500 potential rare disease diagnoses. All 140 individuals lacked molecular diagnoses after routine clinical NGS testing, and by application of ExomeDepth, we identified 17 CNVs contributing to the cause of a Mendelian disorder. Our findings support the integration of CNV detection using ExomeDepth v1.1.6 with routine targeted NGS diagnostic services for Mendelian disorders. Implementation of this strategy increases diagnostic yields and enhances clinical care.
Similar content being viewed by others


Introduction
Molecular diagnostic services available for patients with genetically heterogeneous Mendelian disease have been transformed by the adoption of next-generation DNA sequencing (NGS) within the clinical setting.1, 2 At present, diagnostic services facilitated by NGS are frequently limited to targeted capture techniques, including custom gene panels3, 4 and whole-exome sequencing (WES).5, 6 These techniques have demonstrated tremendous power to identify rare and private single-nucleotide variation and small insertions/deletions underpinning disease onset.
The identification of large structural variants and copy number variants (CNVs) encapsulating the regions targeted by WES and custom gene panel assays have proved challenging in a clinical context. Although whole genome sequencing (WGS) techniques have the potential to address this gap in diagnostic NGS services,7, 8 the cost and data burdens remain substantial. Consequently, the application of CNV detection algorithms in targeted NGS diagnostic services can facilitate immediate improvement in clinical care for individuals with heterogeneous Mendelian disorders. However, such techniques require formal assessment to demonstrate accuracy, reliability and repeatability.
Here we assess a framework for the implementation of CNV detection with targeted NGS diagnostic services applied across a range of highly heterogeneous Mendelian disorders.
Methods
Study design
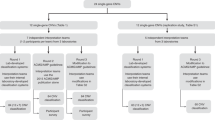
High coverage targeted NGS data was generated in a UK Accredited Clinical Laboratory. We applied a CNV detection algorithm to validate the sensitivity for (i) known CNV events, and (ii) simulated CNV events (Figure 1). We assessed a number of factors to determine whether they influenced successful CNV surveillance. We selected two key factors identified from assessments of simulated and known CNVs (inter-sample variability and insufficient coverage) as quality assurance metrics during prospective CNV detection for individuals without molecular diagnoses through clinical NGS testing (Figure 1).

Study design. The approach taken in this study is to assess the ‘accuracy’ and ‘key factors’ influencing the accuracy of ExomeDepth applied to targeted NGS data sets. The ‘key factors’ assessed by application of ExomeDepth to data sets with known and simulated CNVs are outlined in Table 1. Gene panel, NGS data generated in a diagnostic environment after enrichment for a set of genes known as a cause of specific Mendelian disorders.
Our analyses included individuals referred for diagnostic testing of four highly heterogeneous disorders where targeted gene panel NGS is a routine diagnostic service, specifically: inherited retinal dystrophies (IRD), congenital cataracts, cardiac disorders, and metabolic disorders.
Sequencing and variant analysis
Whole genome and targeted NGS
WGS data was generated for 25 individuals by Complete Genomics (Mountain View, CA, USA) using a mate-paired sequencing technique, as described previously.9 Read alignment and variant calling was performed using version 2.5 of the Complete Genomics pipeline.10
For targeted NGS, enrichments were performed on DNA extracted from peripheral blood using Agilent SureSelect Custom Design target-enrichment kits (Agilent, Santa Clara, CA, USA). Enrichment kits were designed to capture known pathogenic intronic variants and the protein-coding regions +/−50 nucleotides of selected NCBI RefSeq transcripts; conditions tested included IRD (105 genes or 180 genes), congenital cataracts (114 genes), cardiac disorders (72 genes comprised of 10 subpanels) and metabolic disorders (226 genes comprised of 6 subpanels). The genes and transcripts included in the targeted capture regions for each disease referral are available online (Supplementary Tables S1–S4) and through the UK Genetic Testing Network (http://ukgtn.nhs.uk/find-a-test/search-by-laboratory/laboratory/manchester-rgc-36/). Samples were pooled and paired-end NGS was performed using the manufacturer’s protocols for the Illumina HiSeq 2000/2500 platform (Illumina, San Diego, CA, USA). Sequencing reads were demultiplexed with CASAVA v1.8.2. and aligned to the hg19 reference genome using the Burrows–Wheeler Aligner short read (BWA-short v0.6.2) software11 before duplicate reads were removed using samtools v0.1.18. 10.3 million unique NGS reads were generated, on average, per sample (n=170, min.=1 241 785, max.=23 240 481, median=10 812 279), with an average coverage of 880 unique reads per nucleotide (n=70 514 012, min.=0, max.=7956, median=783, SD=515.4) and 2155 unique reads per exon (n=388 974, min.=0, max.=317 678, median=1561, SD=3309.8) within the complete region enriched for analysis. The detection and clinical analysis of single-nucleotide variants and small insertions/deletions was performed as described previously.4, 12
CNV detection
For the 25 samples with WGS data, CNVs were identified using version 2.5 of the Complete Genomics pipeline.10 Briefly, this strategy incorporates an assessment of (i) sequencing read depth, and (ii) discordant mate-pairs. For each tested individual, sequencing read depth was normalized for GC content and genomic positional effects, and CNV status was calculated for non-overlapping 2 Kb genomic intervals through comparison to a baseline sample set—comprised of 52 unrelated individuals. To identify the location of breakpoints and insertion points of CNV events, genomic regions where mate-pairs aligned to the reference genome displayed abnormal genomic intervals between the two reads were flagged. Within these flagged regions, local de novo assembly was then performed for sequencing reads where only one of the two reads within the mate-pair aligned to the reference genome. Where possible, the genomic location of breakpoints and insertion points was identified and reported.
For targeted NGS samples, CNV detection was performed using ExomeDepth v1.1.6.13 For each tested individual, the ExomeDepth algorithm builds the most suitable reference set from the BAM files of a presented group of potential reference samples. We presented ExomeDepth with BAM files for >20 individuals that had been generated by identical laboratory and computational procedures. All potential reference samples were individuals referred for genomic diagnostic testing who were not knowingly related to the tested individual and had been obtained from the same sequencing run on the Illumina HiSeq platform, where possible. The reference sample sets selected by ExomeDepth are referred to as ‘reference samples’ herein.
Accuracy of ExomeDepth in comparison to WGS and multiplex ligation-dependent probe amplification (MLPA)
For 25 individuals with IRD, we generated gene panel NGS and WGS data sets (Figure 1). We used the variant detection techniques applied to the WGS data sets as a reference standard for CNV detection and then assessed, at the exon level (n=1590 exons per sample), the sensitivity and specificity of ExomeDepth applied to gene panel NGS data sets. We defined sensitivity as the capability of ExomeDepth to identify exons with abnormal CNV status and specificity as the capability to identify exons with a normal CNV status. For a further five individuals with cardiac disorders, we generated gene panel NGS and MLPA data sets (Figure 1) and then assessed the sensitivity of ExomeDepth applied to gene panel NGS data sets for these individuals.
Assessment of ExomeDepth to identify simulated CNV events
Simulated CNV events were introduced into targeted NGS data for the 25 IRD patients with complementary WGS data. The enrichment region for targeted NGS for the 25 samples encapsulates 1590 protein-coding exons for 105 genes associated with IRD. Importantly, we had previously defined and reported the copy number status for each exon included within the targeted enrichment through the analysis of WGS data.7
Simulation was performed using a random sample and exon selector, bedtools v2.16.2 intersect and software within the PicardTools v1.75 java package: DownsampleSam and MergeSamFiles (Supplementary Figure S1). Exons were excluded from the analysis if they overlapped with known heterozygous deletion events in the selected sample. We simulated deletion events for 1000 single exons and 1749 multiple exons (2–4 exon events). In all cases, we assume that the intronic breakpoints of the deletion event are not captured through NGS. Deletion events are not expected to be detected above a test:reference sample read ratio of 0.7 (see Supplementary Results and Methods). We created three discrete groups for simulated deletion events, with the extent of sequencing reads randomly removed indicated in parentheses: (i) control events (0%), (ii) deletion with amplification bias (40%), and (iii) deletion without amplification bias (50%). Further details on the simulation methodology are provided in Supplementary Results and Methods.
Assessment of factors influencing successful identification of CNV events
We assessed a number of criteria for known and simulated CNV events in order to assess whether they are key determinants of successful CNV surveillance through ExomeDepth, including: (i) the intra-sample variation in coverage, using the normalized read count (reads-per-kilobase-per-million, rpkm) coefficient of variation (CV) for surveyed genes in test samples (Supplementary Figure S2), (ii) the inter-sample variation in coverage, using the rpkmCV for surveyed exons across reference samples selected by ExomeDepth (Supplementary Figure S2), (iii) the percentage of nucleotides and the number of exons containing nucleotides with appropriate sequencing depth for in-house diagnostic surveillance (>50 × unique sequencing reads), (iv) the total and normalized read depth across surveyed exons, (v) the GC content of the surveyed regions, (vi) the size of exons, and (vii) the distance between neighbouring exons. All statistical analyses were performed in the R v3.2.1 software.
Integration of CNV detection during clinical NGS testing
We integrated CNV detection using ExomeDepth into the NGS workflow for 140 individuals from 12 distinct referral groups (Supplementary Table S5). The reasons for assessment of CNV events were (i) an assessment of whether a heterozygous CNV event was in trans to a clearly or likely pathogenic variant or (ii) an assessment of whether a heterozygous CNV event was present in a gene highly specific to an individual’s clinical presentation. In accordance with the recommendations of the ExomeDepth developers, test samples with an overall correlation to selected reference samples <0.97 were repeated with an alternative set of reference samples or excluded from analysis. Clinical interpretation of CNVs was restricted to genes relevant to their referral on a case-by-case basis. We performed additional assays to confirm the presence of all identified CNVs before they were clinically reported. Where kits designed and created by MRC-Holland (Amsterdam, Netherlands) were available, we carried out MLPA assays. In the absence of a suitable MLPA kit, we validated CNVs using droplet digital PCR or a bespoke multiplex quantitative fluorescence methodology (see Supplementary Methods). Validated CNV events were submitted to the ClinVar database.14
Results
Accuracy of ExomeDepth in comparison to WGS and MLPA
To establish the accuracy and reliability of ExomeDepth when applied to targeted NGS data, we analysed targeted NGS data sets for 30 individuals in whom CNV detection had been performed using either WGS (n=25) or MLPA (n=5). This allowed calculation of the sensitivity and specificity for identified deletions and duplications. Overall, we found a sensitivity of 92.9% and identified that variable and insufficient coverage within surveyed genes reduces the capability of ExomeDepth to identify single-exon deletions.
In comparison to WGS, we determined that ExomeDepth applied to targeted NGS data sets (encompassing 1590 exons from 105 genes) has a sensitivity of 100% and a specificity of 99.8% (Supplementary Table S6) at the exon level. True positive events included a single exon deletion in GPR98, a two-exon deletion in USH2A and a six-exon deletion in PCDH15 (Supplementary Table S7). In comparison to MLPA, we identified three out of the four single-exon deletions and one single-exon duplication (Supplementary Table S7). We assessed a number of key factors and observed that the sequencing data for the individual in whom a single-exon deletion was erroneously not identified showed the highest intra-sample variation (62%) and the highest level of insufficient coverage (9.5% of exons and 0.86% of nucleotides; sample 14011718, Supplementary Table S8).
We assessed metrics calculated by ExomeDepth for the eight previously identified deletions and duplication events, observing that the average confidence (Bayes factor) determined by ExomeDepth for true positive CNV events was 45.04 (Supplementary Table S7, min.=6.4, max.=76.8) and the average ratio of sequencing reads between test and reference samples for deletions was 0.61 (Supplementary Table S7, min.=0.539, max.=0.745) and 1.4 for the sole duplication event.
Capability of ExomeDepth to identify simulated CNV events
In order to assess factors that influence the successful identification of CNV events in targeted NGS data using ExomeDepth, we introduced simulated events, in silico, into the targeted NGS data sets created in a clinical setting for the 25 individuals for whom we held complementary WGS data. We found a 97% sensitivity for simulated events when 50% of the NGS reads were removed from selected exons (n=2749) and identified that inter-sample variation—a measure of consistency of NGS read coverage across reference samples (Supplementary Figure S2)—and insufficient coverage were key determinants of whether simulated events were missed or identified by ExomeDepth (Table 1 and Supplementary Table S9).
Single-exon deletions (n=1000) were introduced into 101 of the 105 genes enriched during NGS and we observed that the sensitivity of ExomeDepth for simulated events was 93.5%, with 930 deletions precisely detected at the exon level and 5 included in deletion events erroneously identified as spanning to adjacent exons. This sensitivity is reduced to 79.5% when accounting for amplification bias in simulated events (Supplementary Tables S10 and S11), with an additional 140 false negative events identified when only 40% of the original NGS reads were removed from the selected exon. Interestingly, 51% (36/70) of the false negative simulated events without amplication bias (50% of NGS reads removed) were exons flanked by neighbouring exons within 250 nucleotides of the canonical donor or acceptor sites. Further, all of these 36 events could be identified if the neighbouring exon boundaries were merged into a single analysis region for simulations, increasing the overall sensitivity of ExomeDepth for simulated events to 97.1% (Supplementary Table S11).
Multiple-exon deletions (n=1749)—where 50% of the NGS reads were randomly removed from adjacent exons—were introduced into all of the 105 genes enriched during targeted NGS for all 25 individuals. We observed sensitivity rates of 96.6% (n=620), 95.9% (n=586) and 97.1% (n=543) for two-exon, three-exon and four-exon deletions, respectively.
To ensure that the process of introducing simulated events into targeted NGS data did not influence the performance of ExomeDepth, we performed the same computational processes of the simulation technique for each event, without removing any NGS reads. No single-exon or multiple-exon simulated deletion events were identified by ExomeDepth in any of these control simulation experiments.
Integration of CNV detection during clinical NGS testing
Following assessment of the accuracy and the reliability of ExomeDepth applied to targeted NGS data sets, we then integrated CNV detection using ExomeDepth into the NGS workflow for 140 individuals from 12 distinct referral groups to assess specific clinical evaluations. These included either (i) an assessment of whether a heterozygous CNV event was in trans to a clearly or likely pathogenic variant, or (ii) an assessment of whether a heterozygous CNV event was present in a gene highly specific to an individual’s clinical presentation. This analysis strategy led to the surveillance of a single gene for 128 individuals, two genes for 10 individuals and three genes for 2 individuals.
Confirmation of molecular diagnoses for 17 individuals
Analysis on a gene-by-patient basis identified 17 heterozygous CNV events (15 deletions, 1 duplication and 1 complex event; Supplementary Table S12; Supplementary Figure S3). All events were verified through an alternative technique, were concluded to contribute to the molecular diagnosis for referred individuals and have been submitted to the ClinVar database (Accession numbers: SCV000493113 - SCV000493128). The heterozygous CNV events identified by ExomeDepth ranged from a 20-exon deletion in PCDH15 (NG_009191.2, NM_001142770.1; >600Kb) to single-exon deletions in RPGRIP1 (NG_008933.1, NM_020366.3), BEST1 (NG_009033.1, NM_004183.3) and NMNAT1 (NG_032954.1, NM_022787.3). For a single individual referred with a provisional clinical diagnosis of Marfan syndrome, we identified a complex event in FBN1 (NG_008805.2, NM_000138.4): a three-exon deletion (chr15:48737523-48741140, c.(5545+1_5546-1)_(5917+1_5918-1)del) and a two-exon duplication (chr15:48720493-48723049, c.(6739+1_6740-1)_(6997+1_6998-1)dup), consistent with a clinical diagnosis of Marfan syndrome (Figure 2).

FBN1 CNV. A complex three-exon deletion, c.(5545+1_5546-1)_(5917+1_5918-1)del, and two-exon duplication, c.(6739+1_6740-1)_(6997+1_6998-1)dup, event identified in FBN1 (NG_008805.2, NM_000138.4), confirming a clinical diagnosis of Marfan syndrome for the referred individual. Crosshairs, the ratio of reads between test and reference samples; grey bar, the 95% confidence interval of expected read ratios in comparison to reference samples.
We assessed metrics calculated by ExomeDepth for identified deletion and duplication events, observing that the average confidence score (Bayes factor) attributed to identified CNV events by the ExomeDepth algorithm was 87 (Supplementary Table S12, min.=22, max.=321) and the average read count ratio between test and selected reference samples was 0.56 (min.=0.518, max.=0.637) and 1.35 (min.=1.31, max.=1.38), respectively.
Accuracy of ExomeDepth applied in a clinical context
To estimate the accuracy of ExomeDepth applied to targeted NGS data sets for the 123 individuals determined to be absent of CNV events, we assessed (i) CNV status through orthogonal techniques, and (ii) two key factors identified through assessments of simulated and known CNV variants: inter-sample variation and insufficient coverage (Table 1).
We calculated the sequencing coverage for each individual and identified that 3% (135/4551) of the surveyed exons contained at least one nucleotide with <50 unique NGS reads. Nine of these exons were found in individuals with a confirmed CNV event in the gene, and 28 were in a gene confirmed to be absent of a CNV event through orthogonal techniques (MLPA; Supplementary Figure S4). Of the remaining 97 exons, 34 were unique patient–exon combinations and 63 were accounted for by 12 exons with insufficient coverage across multiple samples. On average, 4.6% of the nucleotides within these 97 poor coverage exons received <50 unique NGS reads (n=97, min.=0.1%, max.=40.9%, median=3.6%), and all exons were within the range of insufficient coverage values observed for true positive simulated deletion events (Table 1).
To estimate the accuracy of ExomeDepth in relation to reference samples, we calculated the variability of sequencing coverage across the selected references for each individual and identified an average inter-sample variation for surveyed exons of 5.1% (n=4551, SD=3.4%), with average minimum and maximum values observed per-individual of 2.4% (SD=1.9%) and 9.9% (SD=5.5%), respectively. In comparison to simulated single-exon deletions, these data are consistent with an average sensitivity of 98.7% (SD=1.5%, min.=88.7%, max.=100%; Figure 3).

Inter-sample variation in sequencing coverage across surveyed exons. Simulations, the variability of sequencing coverage in selected reference samples for 971 identified and 29 missed single-exon-simulated deletions. Diagnostic survey, the variability of sequencing coverage in selected reference samples for 4551 exons surveyed for CNVs in a diagnostic context.
For six individuals, data from MLPA analyses provided additional support for the absence of a CNV event (Supplementary Figure S4). For a single individual, we identified a false negative event after subsequent MLPA analysis of the DSP gene. We found that alteration of the analysis region-to survey five subexonic regions enriched by non-overlapping probes though ExomeDepth-identified a partial exon duplication event within the DSP gene which complemented the result from MLPA (Supplementary Figure S5).
Discussion
CNVs are an important and common form of genomic variation in the general population15, 16 and are implicated in many Mendelian disorders.7, 8, 17 An ability to accurately survey for CNV events, in particular in targeted NGS data sets, therefore has the power to increase diagnostic yields and enhance clinical care. Although it has already been shown that read-depth CNV detection algorithms can be successfully applied to targeted NGS data in a research context,13, 18, 19, 20 their integration within diagnostic services has been slower due to a lack of validation parameters. In this study, we have identified key factors that can facilitate the successful application of a widely used bioinformatics tool, ExomeDepth,13 for CNV surveillance of targeted NGS data sets within the clinical environment.
CNV detection tools used in a diagnostic context must be able to identify deletion and duplication events that encapsulate single targets/exons included within the targeted enrichments of custom gene panel and WES techniques, which is a known limitation of some publically available algorithms. As large data sets of known true positive single-exon CNV events do not exist, we have developed and applied a computational simulation technique that permits extended assessment of single-exon CNV events. As a result, we have been able to perform an assessment of trends in large and controlled data sets (Table 1), We have then used real-time comparison between WGS and targeted NGS data to assess their applicability to real data sets. Using this combined approach, we have shown that amplification bias within NGS assays and the distance between exons enriched during NGS influences the overall sensitivity of ExomeDepth (Supplementary Table S11). After accounting for these dominating factors, we have demonstrated how variability of sequencing coverage between and within samples, the extent of read depth, the size of surveyed exons and the level of insufficient coverage are important determinants of successful identification of single-exon deletion events through ExomeDepth (Table 1,Supplementary Table S9 and S10). Although all these metrics are indicated as important quality assurance parameters for the accurate detection of single-exon CNVs, they are neither completely independent nor equally applicable to real data sets on an individual basis. We therefore selected two key metrics for routine incorporation into diagnostics: insufficient coverage (test sample dependent) and inter-sample variability (reference sample dependent). This two-part process first checks for the quantity of sequencing coverage over exons surveyed in the tested sample and, second, assesses the consistency of NGS read coverage across reference samples for each surveyed exon. We have assimilated this information to successfully integrate surveillance of CNVs into the clinical bioinformatics pipeline for 140 individuals in a clinical setting, achieving a definitive molecular diagnosis in 17 of the 140 individuals. Importantly, we have shown that 97.2% of the exons surveyed and determined to be absent of a CNV event have sufficient coverage, and none of the insufficiently covered exons lie outside the range of true positives identified from simulated experiments. Moreover, we have calculated the inter-sample variability for surveyed exons on an individual basis, and through comparison to simulated single exon events, estimated the accuracy of ExomeDepth to be 98.7% for the 123 individuals without an identified CNV (Figure 3). Both of these quality assurance observations are supported by their integration with other CNV software tools21 and the absence of CNV events in six individuals tested through MLPA.
Taken together, our data illustrate the utility of CNV assessments within a diagnostic setting using the publically available ExomeDepth software and support the utilization of quality assurance parameters in complement to CNV detection algorithms in targeted NGS diagnostic services. Although other types of software can be routinely applied to WGS data sets to detect CNVs at single-nucleotide resolution, we expect that application of the approaches outlined in this study will improve the utilization of read depth CNV tools in diagnostic environments across heterogeneous targeted NGS gene panel approaches, including small and large gene panels, as described here, and WES.
References
Baetens M, Van Laer L, De Leeneer K et al: Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat 2011; 32: 1053–1062.
O'Sullivan J, Mullaney BG, Bhaskar SS et al: A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet 2012; 49: 322–326.
Nishio SY, Hayashi Y, Watanabe M, Usami SI : Clinical application of a custom AmpliSeq library and ion torrent PGM sequencing to comprehensive mutation screening for deafness genes. Genet Test Mol Biomarkers 2015; 19: 209–217.
Ellingford JM, Barton S, Bhaskar S et al: Molecular findings from 537 individuals with inherited retinal disease. J Med Genet 2016; 53: 761–767.
Lee H, Deignan JL, Dorrani N et al: Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014; 312: 1880–1887.
Yang Y, Muzny DM, Xia F et al: Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014; 312: 1870–1879.
Ellingford JM, Barton S, Bhaskar S et al: Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology 2016; 123: 1143–1150.
Gilissen C, Hehir-Kwa JY, Thung DT et al: Genome sequencing identifies major causes of severe intellectual disability. Nature 2014; 511: 344–347.
Drmanac R, Sparks AB, Callow MJ et al: Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 2010; 327: 78–81.
Carnevali P, Baccash J, Halpern AL et al: Computational techniques for human genome resequencing using mated gapped reads. J Comput Biol 2012; 19: 279–292.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Gillespie RL, O'Sullivan J, Ashworth J et al: Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014; 121: 2124–2137.
Plagnol V, Curtis J, Epstein M et al: A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012; 28: 2747–2754.
Landrum MJ, Lee JM, Benson M et al: ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016; 44: D862–D868.
Iafrate AJ, Feuk L, Rivera MN et al: Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Sebat J, Lakshmi B, Troge J et al: Large-scale copy number polymorphism in the human genome. Science 2004; 305: 525–528.
Glessner JT, Bick AG, Ito K et al: Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 2014; 115: 884–896.
Krumm N, Sudmant PH, Ko A et al: Copy number variation detection and genotyping from exome sequence data. Genome Res 2012; 22: 1525–1532.
Fromer M, Moran JL, Chambert K et al: Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 2012; 91: 597–607.
Li J, Lupat R, Amarasinghe KC et al: CONTRA: copy number analysis for targeted resequencing. Bioinformatics 2012; 28: 1307–1313.
Johansson LF, van Dijk F, de Boer EN et al: CoNVaDING: single exon variation detection in targeted NGS data. Hum Mutat 2016; 37: 457–464.
Acknowledgements
We thank all patients, referring clinicians, clinical scientists and genetic counselors involved in this study. This work was supported by the Biotechnology and Biological Sciences Research Council, the Manchester Biomedical Research Centre, The National Institute for Health Research Biomedical Centre at Moorfields Eye Hospital and the UCL Institute of Ophthalmology, the DST UK-India Education and Research Initiative and an independent research grant funded by the Manchester Academic Health Science Centre.
Author contributions
JME, GCMB, SCR and BP designed and coordinated the study. JME, CC, StB, SaB, SG, RLG, PIS, BH, MM, ARW, WGN, SCR and GCMB contributed genetic and/or phenotypic data. JME wrote the manuscript and all authors provided important revisions and intellectual content.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ellingford, J., Campbell, C., Barton, S. et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur J Hum Genet 25, 719–724 (2017). https://doi.org/10.1038/ejhg.2017.42
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.42
This article is cited by
-
Detection of the Copy Number Variants of Genes in Patients with Familial Cardiac Diseases by Massively Parallel Sequencing
Molecular Diagnosis & Therapy (2023)
-
Clinical and technical assessment of MedExome vs. NGS panels in patients with suspected genetic disorders in Southwestern Ontario
Journal of Human Genetics (2021)
-
Next-generation gene panel testing in adolescents and adults in a medical neuropsychiatric genetics clinic
neurogenetics (2021)
-
Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes
Scientific Reports (2021)
-
Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort
European Journal of Human Genetics (2021)
