
Abstract
Recent research has suggested that transmembrane protein 65 (TMEM65) is localized within the inner mitochondrial membrane. Little else is known about its function. In this study we investigated the location and function of TMEM65. Further, we report the functional consequences of a novel homozygous splice variant (c.472+1G>A) in the TMEM65 gene in a patient with mitochondrial encephalomyopathy. Here we investigated the location of TMEM65 by immunofluorescence staining of the protein and by immunoblotting of the isolated mitochondrial fractions in healthy fibroblasts and those from the patient. To study the function of TMEM65 we knocked down mRNA using TMEM65-specific siRNA, and measured mitochondrial function by enzymology, protein abundance and oxygen consumption rate in fibroblasts. Subcellular fractionation confirmed that the TMEM65 protein was present in the inner mitochondrial membrane. Knocking down TMEM65 expression in dermal fibroblasts severely affected mitochondrial content and respiration rate. Further evidence for the essential role of TMEM65 in mitochondrial function came from the demonstration of severe cellular and clinical consequences resulting from the novel TMEM65 gene mutation. In conclusion, these findings suggest that TMEM65, an inner mitochondrial membrane protein, plays a significant role in mitochondrial respiratory chain function. We also provide the first evidence that a mutation in the TMEM65 gene results in mitochondrial dysfunction and a severe mitochondrial encephalomyopathy phenotype.
Similar content being viewed by others

Introduction
Mitochondrial cytopathies are common inborn errors of metabolism estimated to affect approximately 1 in 6000 people.1 Molecular diagnosis can be challenging due to factors such as the dual genomic contribution to mitochondrial transcripts by mitochondrial DNA-encoded and nuclear DNA-encoded genes, and the frequent overlap of clinical phenotypes with many other genetic and non-genetic disorders. Most of the disease-causing mutations responsible for mitochondrial cytopathies were first identified in the relatively small (16.5 kb) mitochondrial DNA;2, 3 however, an increasing number of pathogenic mutations in nuclear-encoded mitochondrial genes are being characterized with the advent of modern sequencing technologies.4, 5, 6 Next-generation exome sequencing will continue to reveal novel genetic variants and mutations that will expand our understanding of disease causality as well as accurately infer the incidence of specific diseases in the population.7, 8, 9
To date, only one mitochondrial transmembrane protein, transmembrane protein 70 (TMEM70), has been implicated as a cause of primary mitochondrial cytopathy (OMIM 612418). Native TMEM70 is involved in the assembly of mitochondrial complex V (ATP synthase).6 Patients with disease-causing TMEM70 mutations show multisystem deficiency in ATP synthase, and usually present with hypertrophic cardiomyopathy, hypotonia, lactic acidosis and 3-methyl glutaconic aciduria.6 Some patients also have dysmorphic features, psychomotor regression, intrauterine growth retardation and gastrointestinal dysfunction.10
A disease that can indirectly influence another mitochondrial transmembrane protein (TMEM65) is French–Canadian Leigh Syndrome caused by mutations in the gene encoding leucine-rich pentatricopeptide repeat-containing protein (LRPPRC).11 LRPPRC associates with the Stem–Loop-Interacting RNA-binding Protein (SLIRP), forming a ribonucleoprotein complex. The Steroid Receptor RNA Activator (SRA), a long non-coding RNA that regulates the activity of nuclear receptors, associates with SLIRP and regulates downstream target genes, including TMEM65.13 The finding that the TMEM65 protein is associated with the inner mitochondrial membrane (IMM),12 and that it is regulated by human SRA gene,13 suggests that TMEM65 likely regulates some aspects of mitochondrial function. Until now, a clinical phenotype resulting from mutations in the TMEM65 gene has not been documented. We report herein a patient with a complex encephalomyopathic phenotype and suspected mitochondrial disease resulting from a homozygous splice-site mutation in the TMEM65 gene identified through whole-exome sequencing (WES). We provide in vivo and in vitro support that the TMEM65 variant is indeed a pathogenic mutation.
Materials and methods
Genotyping and linkage analysis
WES was performed as a part of the FORGE (Finding Of Rare disease GEnes) Canada Consortium/Care4Rare Canada Consortium research initiative.14 Approximately 97% of exons were covered at >10 ×. To identify likely causal variants, we filtered variants seen in the proband to retain only those that were (a) homozygous in the proband, (b) not homozygous in the proband's unaffected sibling, (c) seen at less than 3% minor allele frequency in both 1000 genomes and the NHLBI Exome Sequencing Project and (d) seen in fewer than 6 of ~200 control exomes sequenced by the McGill University and Genome Quebec Innovation Centre. All genotypic and phenotypic data for this project, including this one, has been uploaded into PhenomeCentral (https://www.phenomecentral.org), a controlled-access database available to clinicians and researchers within the rare disease community. The research study was approved by the McMaster University Research Ethics Board (REB# 11-427-T). The patient’s parents signed an approved informed consent document as a part of enrollment. Follow-up confirmatory testing in the proband and parents was performed using Sanger sequencing of amplified TMEM65.
Cell culture and reagents
A primary fibroblast culture was established from a skin biopsy of the patient. Biopsies were performed with written informed consent and in accordance with protocols approved by the Research Ethics Board at McMaster University.
siRNA knockdown
Small interfering RNAs (siRNAs; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) were used for silencing the expression of hTMEM65 mRNA in dermal fibroblasts from age-matched healthy controls. As a negative control, non-silencing siRNA (Scrambled siRNA-A; Santa Cruz Biotechnology, Inc.) was used. Transfection was performed using siRNA transfection reagent (Santa Cruz Biotechnology, Inc.) according to the manufacturer’s instructions.
TMEM65 expression plasmid complementation
For transfection of the TMEM65 cDNA clone plasmid (OriGene Technologies, Rockville, MD, USA) into dermal fibroblasts, Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used according to the manufacturer's instructions.
TMEM65 immunofluorescence staining
For TMEM65 immunofluorescence staining, the fibroblasts were cultured on chamber slides and fixed. Cells were stained with primary TMEM65 rabbit polyclonal antibody (K-15: sc87463; Santa Cruz Biotechnology, Inc.). Then the cells were added with secondary goat anti-rabbit IgG Alexa Fluor 488-conjugated antibodies (Abcam, Cambridge, MA, USA). Images were visualized using a confocal laser scanning microscope (Olympus, Model IX3-ZDC, Tokyo, Japan), and analyzed by imaging software (Olympus Fluoview version 4.2 software).
Mitochondrial staining
Mitochondrial staining was performed to assess the distribution of mitochondria in cells. Fibroblasts were incubated with the cell-permeant mitochondria-specific red fluorescent probe MitoTracker Deep Red at a final concentration of 500 nm. Cells were washed twice with PBS and fixed with 4% paraformaldehyde. Finally, cells were washed with PBS and immunostained for TMEM65 and mounted with hard set Vecta shield mounting fluid (Vector Laboratories, Inc., Burlington, ON, Canada). Samples were examined under a confocal microscopy using laser scanning microscopy.
Measurements of oxygen consumption rate by a Seahorse XF24 analyzer
Oxygen consumption rate (OCR) was measured in adherent primary dermal fibroblasts with a XFe 24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA). Each control and patient fibroblast cell lines were seeded in quadruplicate wells of a XF24-well cell culture plate. OCR was measured while cells were subjected in sequence to the following additions: (1) basal levels were measured with no additives; (2) 1 μ m oligomycin, (3) 2 μ m FCCP (4) rotenone 1 μ m and (5) antimycin 1 μ m.
Enzyme activities
Cell pellets were re-suspended in potassium phosphate buffer and snap frozen and thawed three times for cell lysis and finally subjected to sonication before enzyme measurements. Mitochondrial electron transport chain cytochrome c oxidase (COX) activity (indicative of mitochondrial oxidative capacity) was determined as previously described by our group.15, 16,17 Citrate synthase activity was assessed as described previously by our group.15, 16 Respiratory chain enzyme analysis on muscle biopsy samples were completed using previously described methods.15–17
mtDNA copy number analysis
Total DNA (genomic and mtDNA) was isolated from dermal fibroblast using the QIAamp DNA Mini kit (Qiagen, Toronto, ON, Canada) according to the manufacturer's instructions. mtDNA copy number, relative to the chromosomal DNA content, was quantitatively analyzed in DNA isolated from fibroblasts using ABI 7300 real-time PCR (Applied Biosystems, Life Technologies Inc., Burlington, ON, Canada). Primers were designed within the ND1 region of the mitochondrial genome. Nuclear β2-globin gene was used as the nuclear marker gene to standardize mtDNA copy number to nuclear DNA content.18
Real-time quantitative PCR
Total RNA was isolated from cell pellet using a total RNA isolation kit in accordance with the manufacturer’s instructions (Qiagen). RNA samples for real-time PCR analyses were reverse-transcribed and quantitative real-time PCRs were performed on the cDNA in the presence of fluorescent dye (SYBR green; Bio-Rad, Mississauga, ON, Canada). Relative expression levels were determined using the comparative cycle threshold method. Primers used for this study are listed in Supplementary Table S2.
Mitochondrial subfractionation
Mitochondria were isolated from quadriceps femoris of C57BL/6 mice using differential centrifugation as previously described.19 Isolated mitochondrial pellets were solubilized and subfractionated into outer mitochondrial membrane (OMM), intermembrane space (IMS), IMM and matrix (Mx) using phosphate swelling-shrinking as previously described.18
Western blotting
Mitochondrial subfractionations and cell homogenates from an age- and sex-matched healthy control and TMEM65 patient were solubilized in RadioImmune Precipitation Assay buffer and were resolved on 12% SDS-PAGE gels. The gels were transferred onto Hybond ECL nitrocellulose membranes (Amersham Biosciences, Mississauga, ON, Canada). Immunoblotting was carried out using TMEM65 (K-15; Santa Cruz), voltage-dependent anion channel (VDAC; Abcam, Toronto, ON, Canada), apoptosis-inducing factor (AIF; Cell Signaling), cytochrome c oxidase subunit I (COX I; Abcam, Canada), superoxide dismutase 2 (SOD2; Abcam, Canada) and pan-actin (Cell Signaling). Membranes were then incubated with anti-rabbit or anti-mouse horseradish peroxidase-linked secondary antibody (GE Healthcare Biosciences, Mississauga, ON, Canada) and were visualized by enhanced chemiluminescence detection reagent (Amersham Biosciences).
Statistical methods
The data obtained were analyzed by either one-way ANOVA or two-way ANOVA followed by the Bonferroni multiple comparison test for multiple group analysis. The P-value <0.05 was considered as significant in all variables. The data were analyzed and represented as mean±SEM.
Results
History and clinical phenotyping of patient
The proband was referred for neurometabolic consultation at 3 years of age with new onset seizures and developmental regression occurring on a background history of microcephaly, mutism and global developmental delay. She was born at full-term to consanguineous parents (first cousins) of Pakistani descent (Figure 1A). The pregnancy was complicated by an antenatal diagnosis of IUGR and microcephaly. Developmental and gross motor delays were noted within the first year of life. She first began to walk independently at the age of 2.5 years but did not develop a pincer grasp and was reported to always have poor coordination. The seizures were initially right-sided focal seizures with secondary generalization. Prior to the onset of seizures, she was able to roll, sit, stand and walk without support and spoke one or two words clearly. Clinical examination revealed microcephaly and dysmorphic features including low set ears, a low hairline and mild micrognathia with polydactyly and syndactyly of the fifth and sixth toe of the right foot. There was also brachydactyly and clinodactyly of the fourth and fifth digit of the left foot. She was mute and blind with roving eye movements and cannot roll or sit unassisted. Spasticity of the legs (clonus) and dystonic posturing of the arms and legs were also noted on examination.

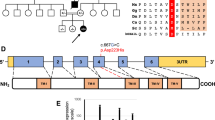
Identification and functional analysis of a novel variant in TMEM65 gene. (A) Pedigree of the family. (B) Electropherogram identifying the heterozygous variant in parents and homozygous variant in the exon 4/intron 4 boundary (NM_194291.2:c.472+1G>A,chr8:g.124,323,320C>T (GRCh38[hg38])) in patient DNA. (C) Real-time PCR of TMEM65 mRNA expression of exon 2 and exon 6 in control (black bars) and in TMEM65 patient (gray bars). The control showed expression of exon 2 and exon 6 while the subject showed lack of mRNA expression of exon 6. (D) Dermal fibroblast from healthy control and TMEM65 patient was cultured and stained for TMEM65. Punctate TMEM65 staining was observed in healthy fibroblast but fibroblast from the TMEM65 patient showed no staining. (E) TMEM65 protein content in control vs TMEM65 patient cells assessed by immunoblotting whole dermal fibroblast cell lysates. The graph shows densitometry measurement of reduced band intensity ratio of TMEM65 to actin in the patient as compared with a healthy age- and sex-matched control. (F) Ultrastructure of skeletal muscle from the TMEM65 patient. a: Mitochondria showed significant pleomorphism and electron densities. b: Some mitochondria contained prominent lipid droplets (white arrow) with most showing electron densities (black arrows). c and d: Sub-sarcolemmal accumulations of rounded mitochondria were frequently seen.
Serology showed normal plasma amino acids and intermittently high lactates (0.9–3.4; normal range=0.5–2.2 mmol/l). MRI of brain at three and a half years of age showed left mesial temporal sclerosis, a mild to moderate volume loss, and a small lactate peak in the bilateral basal ganglia. Follow-up MRI at 8 years of age shows progression of the supra and infratentorial brain atrophy, as noted previously. A new abnormally high T2/FLAIR signal involving cerebral white matter, left cerebellum and thalami was observed, along with significant volume loss of the thalami. At age 10 years she has since developed spasticity and sustained clonus in both feet and is mute, blind and deaf and experiencing 10–15 multi-focal seizures per day in spite of several antiepileptic drug therapies. Unlike TMEM70 mutations, the patient did not show any evidence of cardiomyopathy by echocardiography at that age.
WES variant analyses and mutated TMEM65 expression profiling
After filtering the WES data, 13 candidate variants remained (Supplementary Table S1), one of which was a homozygous variant at the TMEM65 exon 4/intron 4 splice site in the proband (NM_194291.2:c.472+1G>A,chr8:g.124,323,320C>T (GRCh38[hg38]) or g.125335561C>T (GRCh37[hg19]), exons numbered as in ENST00000297632.7). The variant data were submitted to LOVD3.0 (#0000150029; http://databases.lovd.nl/shared/variants/0000150029). As TMEM65 was the only gene with a mitochondrial localization (www.broadinstitute.org/files/shared/metabolism/mitocarta/human.mitocarta2.0.html) and the only variant that had not been reported in a public database, we considered this to be the likely causal variant and pursued validation and further functional studies. The variant was independently confirmed by Sanger sequencing and family studies confirmed that both parents were heterozygous for the variant (Figure 1B) and the variant was not seen in either unaffected sibling (data not shown). Owing to the fact that this was a splice-site variant that would disrupt the canonical GU donor site, we examined exon-specific TMEM65 mRNA expression in patient’s dermal fibroblasts to identify if this variant resulted in a loss of downstream exons in the mRNA transcript. Compared with an age-matched healthy control, dermal fibroblasts from the patient showed no expression of TMEM65 exon 6 while expression for exon 2 was maintained, confirming that the splicing variant altered the integrity of the full-length mRNA product (Figure 1C). The TMEM65 immunofluorescence staining (Figure 1D) and immunoblot (Figure 1E) analysis further confirmed the mRNA expression findings with severely reduced abundance of TMEM65 (25 kDa) protein in patient’s fibroblasts vs a healthy control.
Mitochondrial DNA copy number, microscopy and mitochondrial activity
Mitochondrial DNA copy number in skeletal muscle biopsy tissue from the patient was found to be significantly lower (P<0.0001) than controls (Supplementary Figure S1A). Muscle respiratory chain enzyme analysis revealed significant deficiencies in complex I+III, II+III and complex IV activity (Supplementary Figure S1B). Citrate synthase activity was higher in patient vs control, likely due to an increase in mitochondrial content to compensate for the loss in function. Light microscopic examination of the muscle biopsy was normal with no evidence of ragged red fibers or COX-negative fibers. In contrast, ultrastructural evaluation of the muscle biopsy showed pleomorphic mitochondrial proliferation with elevated lipid droplets and several mitochondria with dark electron densities (Figure 1F).
Intramitochondrial localization of TMEM65 protein
TMEM65 has been reported in MitoCarta as being present in the mitochondria (http://www.broadinstitute.org/files/shared/metabolism/mitocarta/human.mitocarta2.0.html). Recently, TMEM65 was shown to be a mitochondrial-specific integral membrane protein that specifically localized to the IMM.12 We confirmed mitochondrial localization of TMEM65 by double immunostaining healthy control subject dermal fibroblasts with an hTMEM65 antibody and a mitochondrial-targeted stain and showing some degree of co-localization (Figure 2a). The lack of complete co-localization is likely due to the fact that a small fraction of TMEM65 is present in cytosol where it gets translated and/or there is a portion that is in the cytosol as part of the N-terminal mitochondrial targeting sequence containing peptide in transit during shuttling to mitochondria. To better localize the intramitochondrial localization of TMEM65, mitochondria were subfractionated into OMM, IMS, IMM and Mx fractions. These fractions were subsequently immunoblotted for the compartment-specific proteins, including, voltage-dependent anion channel (VDAC, OMM), AIF (IMS), COX I (IMM) and SOD2 (Mx) (Figure 2b). TMEM65, like COX I, is distinctly present in IMM fraction, which supports earlier work also showing TMEM65 localization to the mitochondria and specifically to the IMM.12

TMEM65 localizes to the inner mitochondrial membrane and a mutation in TMEM65 effects cellular respiration. (a) Dermal fibroblasts taken from healthy control were stained for TMEM65 protein (green) and mitochondria (red) and images were taken on an inverted confocal microscope (Olympus). Co-localization of TMEM65 protein (green) and Mitotracker deep red was captured and calculated by Olympus Fluoview software Version 4.2 (scale bar=10 μm). The yellow color is reflective of the co-localization. (b) Mitochondria isolated from skeletal muscle were subfractionated into the outer mitochondrial membrane (OMM), intermembrane space (IMS), inner mitochondrial membrane (IMM) and matrix (Mx) fractions, and these fractions were subsequently immunoblotted for the compartment-specific proteins voltage-dependent anion channel (VDAC), apoptosis-inducing factor (AIF), cytochrome c oxidase subunit I (COX I), transmembrane protein 65 (TMEM65) and superoxide dismutase 2 (SOD2). (c) Oxygen consumption rate in primary dermal fibroblasts from a TMEM65 patient (gray bars) and compared with primary dermal fibroblast from three healthy age-matched controls (black bars). Oxygen consumption rate (OCR) was determined using the XFe24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA, USA). During oxygen consumption rate measurements cells were exposed to metabolic inhibitors including oligomycin, FCCP, rotenone and antimycin. The dermal fibroblast from the TMEM65 patient showed an overall reduced cellular respiration rates. The values are normalized to the total cellular protein per well. Graph represents three individual experiments. **P<0.001, ***P<0.0001.
The TMEM65 variant reduces mitochondrial oxidative capacity
To further assess the impairment in mitochondrial function, we measured oxygen consumption rates in fibroblasts from the patient and healthy subjects using a mitochondrial metabolic flux assay. Compared with healthy subjects, dermal fibroblasts from patient had impaired basal oxygen consumption rate, concomitantly with lower maximal mitochondrial respiration (Figure 2c). Furthermore, mtDNA copy number, CS activity and COX activity were lower in patient fibroblasts as compared with age- and sex- matched healthy controls (Figure 3b).

siRNA-mediated knockdown of TMEM65 in healthy human dermal fibroblast affected mitochondrial function. (a) Oxygen consumption rate was estimated in fibroblasts transfected with control scrambled siRNA (black bars) and TMEM65 siRNA (gray bars). The fibroblasts knocked down for TMEM65 showed significantly lower cellular respiration at baseline, after oligomycin exposure and after FCCP injection when compared with control fibroblasts transfected with scrambled siRNA. Graph represents three individual experiments with different fibroblast cells from three healthy individuals. (b) Patient and TMEM65 siRNA knocked down fibroblasts showed significantly reduced mitochondrial DNA copy number and reduced enzyme activity as shown in representative graph. ***P<0.0001.
Ablation of TMEM65 expression induces mitochondrial OXPHOS dysfunction and lower mtDNA copy number in healthy fibroblasts
We hypothesized that a reduction in TMEM65 expression caused mitochondrial dysfunction. To establish this causal relationship between the reported novel TMEM65 variant with reduced TMEM65 expression and mitochondrial dysfunction, we knocked down TMEM65 expression in fibroblasts from healthy subjects using TMEM65 siRNA to assess if mitochondrial dysfunction ensued. We confirmed effective reduction in TMEM65 mRNA in the cells using RT-PCR (Supplementary Figure S2A). Similar to the effects of variant TMEM65 in patient’s fibroblasts, healthy cells treated with TMEM65 siRNA-mediated knockdown showed similar mitochondrial dysfunction, characterized by impaired basal oxygen consumption rate and reduced maximal mitochondrial respiration compared with scrambled siRNA control (Figure 3a). TMEM65 knockdown also resulted in lower mtDNA copy number and reduced COX and CS activity as compared with scrambled siRNA control in healthy cells, which was consistent with values obtained in fibroblasts from the affected proband (Figure 3b). The reduction in mitochondrial function appears to be global and not subunit specific in that both CS and COX activity were reduced by similar amounts leading to no effect on the COX/CS ratio.
TMEM65 complementation rescues mitochondrial dysfunction and reverses reduction in mtDNA copy number in TMEM65 mutant patient fibroblasts
If the TMEM65 variant was indeed leading to mitochondrial pathology, we hypothesized that overexpression of native human TMEM65 in patient dermal fibroblasts would rescue the inherent mitochondrial OXPHOS deficits. Dermal fibroblasts from the patient that were transfected with a TMEM65 cDNA clone plasmid (OriGene Technologies) and showed: (i) rescue of defective mitochondrial respiration (Figure 4a), (ii) reversal of the mtDNA copy number deficit (Figure 4b) and (iii) normalization of COX and CS activity, back to healthy control levels (Figure 4b). Collectively, the above data establish a strong causal relationship between the novel TMEM65 variant and mitochondrial dysfunction and thus should be considered a true TMEM65 mutation.

TMEM65 complementation in patient fibroblast improves mitochondrial function. (a) The representative graph of oxygen consumption rate (OCR) in healthy control fibroblast (black bars) with TMEM65 patient (gray bars) and compared with patient fibroblast transfected TMEM65 expression vector (open bars). The fibroblast from patient showed significantly reduced cellular respiration at baseline, after Oligomycin exposure and after FCCP injection while the oxygen consumption rate was improved in patient fibroblast transfected with TMEM65 expression plasmid. *P<0.01, **P<0.001, ***P<0.0001. (b) Fibroblasts from control and patient were transfected with TMEM65 expression vector and compared for mtDNA copy number and mitochondrial enzyme activity. Enzyme activity and mtDNA copy number were significantly improved in patient after overexpression of TMEM65. *P<0.01, **P<0.001, ***P<0.0001.
Loss of TMEM65 function results in reduction in mitochondrial gene transcripts and upregulation of hypoxia signaling
Having established that the TMEM65 variant, or siRNA-mediated reduction in TMEM65 mRNA abundance, resulted in mitochondrial dysfunction, we sought to elucidate other aspects of the underlying effects of this observation upon cellular function with a focus on mitochondrial genes and hypoxia signaling. Indeed, TMEM65 knockdown (Supplementary Figure S2A) lowered basal and maximal mitochondrial respiration (Supplementary Figure S2B). This occurred in tandem with upregulation of gene transcripts involved in regulating hypoxia (Supplementary Figure S2C). Additionally, chemical activation of hypoxia by DMOG treatment induced a similar reduction in transcripts of mitochondrial proteins, along with increase in HIF-1α-mediated hypoxia signaling.
Discussion
A previous report suggested that TMEM65 may participate in the regulation of mitochondrial function;12 however, a mutation in TMEM65 and its relation to mitochondrial pathology has not yet been described. Herein, we present a case study of a patient with a homozygous TMEM65 splice-site variant (NM_194291.2:c.472+1G>A, chr8:g.124,323,320C>T (GRCh38[hg38])) and the relationship of the variant to tissue and cellular mitochondrial pathology, establishing it as a pathogenic mutation.
This variant was selected from the list of candidates obtained by WES in the proband for further functional studies because of its previous association with the mitochondria and because it was the only one not previously reported in a public database. According to the Exome Aggregation Consortium (ExAC), variations in TMEM65 are extraordinarily rare and land 2 standard deviations above average for gene constraint, suggesting that the gene is more intolerant to nonsynonymous variation than average (z=2.01). TMEM65 also appears to be relatively intolerant of loss of function variants (pLI=0.59). Only one other loss of function variant was listed in ExAC, a heterozygous stop-gain alteration seen in an individual of European (Non-Finnish) ancestry.
Functional studies for the c.472+1G>A variant seen in the patient reported here showed that this was indeed a mutation in that it caused alterations in downstream mRNA abundance and a severe loss of TMEM65 protein and mitochondrial dysfunction. We have confirmed that TMEM65 resides in IMM, and its expression is important for mitochondrial respiration and mtDNA copy number maintenance. Finally, we have shown that reduction in TMEM65 mRNA abundance (siRNA knockdown) resulted in impaired mitochondrial gene expression, mtDNA depletion and concomitant upregulation of hypoxia signaling.
Many nuclear mutations have now been discovered using WES/next-generation technology that result in primary mitochondrial disease.19, 20, 21, 22 To our knowledge, this is the first report that investigates the functional consequences of a mutation in the TMEM65 gene. The data show that disruption of mRNA with a resultant reduction in TMEM65 protein abundance leads to mitochondrial dysfunction with severe neurological manifestations, including intrauterine growth retardation, dysmorphic features, microcephaly, lactic acidosis, developmental delay with psychomotor regression, intractable seizures, dyskinetic movements, without cardiomyopathy.
The molecular mechanisms regulating aspects of cellular metabolism in relation to a severe reduction in TMEM65 protein abundance are unknown. However, we did show an interesting link between the reduction in TMEM65 protein abundance and upregulation of HIF-1α-mediated hypoxia signaling and impaired mitochondrial respiration. We hypothesize that similar pathways may be affected in cancer metabolism where hypoxia occurs (especially in solid tumors), which results in reduction in mitochondrial respiration and induction of glycolytic pathways (Warburg effect) that are the cornerstone of metastatic tumors. It remains to be elucidated if the significant mitochondrial dysfunction from a severe reduction in TMEM65 protein leads to other cellular consequences linked to mitochondrial pathology, including telomere shortening, oxidative stress, apoptosis and inflammasome activation.
In summary, a mutation in the TMEM65 gene is associated with a severe encephalomyopathic mitochondrial disease phenotype and mutations in this gene should be considered in children with developmental delay, seizures and abnormal skeletal muscle ultrastructural changes.
References
Schaefer AM, McFarland R, Blakely EL et al: Prevalence of mitochondrial DNA disease in adults. Ann Neurol 2008; 63: 35–39.
Wallace DC, Singh G, Lott MT et al: Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 1988; 242: 1427–1430.
Wallace DC, Zheng XX, Lott MT et al: Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 1988; 55: 601–610.
Kirby DM, Salemi R, Sugiana C et al: NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest 2004; 114: 837–845.
Loeffen J, Smeitink J, Triepels R et al: The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet 1998; 63: 1598–1608.
Cizkova A, Stranecky V, Mayr JA et al: TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet 2008; 40: 1288–1290.
Carroll CJ, Brilhante V, Suomalainen A. : Next-generation sequencing for mitochondrial disorders. Br J Pharmacol 2014; 171: 1837–1853.
Sambuughin N, Liu X, Bijarnia S et al: Exome sequencing reveals SCO2 mutations in a family presented with fatal infantile hyperthermia. J Hum Genet 2013; 58: 226–228.
Galmiche L, Serre V, Beinat M et al: Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat 2011; 32: 1225–1231.
Spiegel R, Khayat M, Shalev SA et al: TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: further delineation of a new syndrome. J Med Genet 2011; 48: 177–182.
Gohil VM, Nilsson R, Belcher-Timme CA et al: Mitochondrial and nuclear genomic responses to loss of LRPPRC expression. J Biol Chem 2010; 285: 13742–13747.
Nishimura N, Gotoh T, Oike Y et al: TMEM65 is a mitochondrial inner-membrane protein. Peer J 2014; 2: e349.
Foulds CE, Tsimelzon A, Long W et al: Research resource: expression profiling reveals unexpected targets and functions of the human steroid receptor RNA activator (SRA) gene. Mol Endocrinol 2010; 24: 1090–1105.
Beaulieu CL, Majewski J, Schwartzentruber J et al: FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet 2014; 94: 809–817.
Safdar A, Hamadeh MJ, Kaczor JJ et al: Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One 2010; 5: e10778.
Parise G, Brose AN, Tarnopolsky MA. : Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp Gerontol 2005; 40: 173–180.
McKenzie S, Phillips SM, Carter SL et al: Endurance exercise training attenuates leucine oxidation and BCOAD activation during exercise in humans. Am J Physiol Endocrinol Metab 2000; 278: E580–E587.
Safdar A, Little JP, Stokl AJ et al: Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem 2011; 286: 10605–10617.
Haack TB, Danhauser K, Haberberger B et al: Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet 2010; 42: 1131–1134.
Falk MJ, Shen L, Gonzalez M et al: Mitochondrial Disease Sequence Data Resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol Genet Metab 2015; 114: 388–396.
Pierce SB, Chisholm KM, Lynch ED et al: Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 2011; 108: 6543–6548.
Lines MA, Jobling R, Brady L et al: Peroxisomal D-bifunctional protein deficiency: three adults diagnosed by whole-exome sequencing. Neurology 2014; 82: 963–968.
Acknowledgements
This study was supported by a CIHR grant and a donation from Dan Wright and family and Terry’s and Mario’s Annual Charity Golf Tournament to MAT. A Safdar held a CIHR Banting Postdoctoral Fellowship at the time of the data collection. A Saleem held a NSERC Postdoctoral Fellowship at the time of data collection. We would also like to thank the family for permitting the investigations. Genetic testing was performed under the Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec and Children’s Hospital of Eastern Ontario Foundation. We wish to acknowledge the contribution of the high throughput sequencing platform of the McGill University and Génome Québec Innovation Centre, Montréal, Canada. Acknowledgments also go to the Care4Rare Consortium: ‘Enhanced Care for Rare Genetic Diseases in Canada’; Gene Discovery Steering Committee – Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa), David Dyment (University of Ottawa) as well as Clinical Coordinator Taila Hartley and Project Manager Chandree Beaulieu at the Children’s Hospital of Eastern Ontario Research Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Nazli, A., Safdar, A., Saleem, A. et al. A mutation in the TMEM65 gene results in mitochondrial myopathy with severe neurological manifestations. Eur J Hum Genet 25, 744–751 (2017). https://doi.org/10.1038/ejhg.2017.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.20
This article is cited by
-
TMEM65 promotes gastric tumorigenesis by targeting YWHAZ to activate PI3K-Akt-mTOR pathway and is a therapeutic target
Oncogene (2024)
-
Co-fractionation–mass spectrometry to characterize native mitochondrial protein assemblies in mammalian neurons and brain
Nature Protocols (2023)
-
Panoramic variation analysis of a family with neurodevelopmental disorders caused by biallelic loss-of-function variants in TMEM141, DDHD2, and LHFPL5
Frontiers of Medicine (2023)
-
Functional characterization of variants of unknown significance in a spinocerebellar ataxia patient using an unsupervised machine learning pipeline
Human Genome Variation (2022)
-
The chromatin remodeler CHD6 promotes colorectal cancer development by regulating TMEM65-mediated mitochondrial dynamics via EGF and Wnt signaling
Cell Discovery (2022)
