
Abstract
Nager syndrome is a rare preaxial acrofacial dysostosis that is caused by heterozygous loss-of-function variants in SF3B4. This gene encodes for a protein required for the assembly of spliceosomal complexes, being a master gene for splicing regulation. The main clinical features of Nager syndrome include facial-mandibular and preaxial limb malformations, with normal cognitive functioning. Most Nager patients are sporadic, but few familial cases with a highly variable phenotype have been reported. In this work, we report a novel synonymous variant within exon 3 of the SF3B4 gene in a family with three members affected by Nager syndrome. No pathogenic variants have been detected in other 24 genes associated with syndromes characterized by mandibulo-facial anomalies. The pathogenicity of the mutation was demonstrated through a hybrid minigene assay, which confirmed an aberrant splicing with the creation of a cryptic splice site, and showed that this allele is hypomorphic. Our findings emphasize the importance to perform functional analyses to assess the possible consequences of synonymous variants and confirmed that hybrid minigenes represent an effective tool to evaluate the effects of variants on splicing, particularly when RNA is not available.
Similar content being viewed by others
Introduction
Nager syndrome (preaxial acrofacial dysostosis; MIM #154400) is a rare, autosomal dominant, genetic disease characterized by facial anomalies and preaxial limb malformations. Most cases are sporadic; however, multiplex families showing a highly variable expressivity of the disease have been reported so far.1, 2, 3
Haploinsufficiency of SF3B4, a gene encoding for a protein involved in the assembly of spliceosomal complexes, is the only known cause of Nager syndrome, that affects greater than 50% of clinically diagnosed patients.4, 5 Since 2012, heterozygous loss-of-function SF3B4 variants have been reported in 38 out of 62 pedigrees with a clinical diagnosis of this syndrome, suggesting genetic heterogeneity;4, 5, 6, 7, 8, 9 in addition, deletions encompassing SF3B4 have been detected in cases with clinical features of Nager syndrome.9, 10
In this article, we describe a three-generation family with Nager syndrome due to a SF3B4 synonymous variant leading to abnormal splicing of the primary transcript, as assessed through a hybrid minigene assay.
Subjects and methods
Case reports
The propositus (P1) is a 3-year-old Caucasian male who was referred to our institute for the evaluation of craniofacial and limb anomalies. He was born at term after prenatal diagnosis of severe micrognathia and polyhydramnios. Birth weight was 3480 g (50th centile), length was 49 cm (10th–25th centile) and head circumference was 34 cm (25th centile). At birth, severe micrognathia, submucous cleft palate and bilateral thumb hypoplasia were observed (Figure 1c and d).

Craniofacial and limb findings in P1 and P2. (a) Facial features of P1 at 5 months include down-slanting palpebral fissures, malar hypoplasia and micrognathia. (b) Floating left thumb of P1. (c, d) Upper limbs radiographs of P1 showing a mildly hypoplastic left radius with absent first metacarpo-phalangeal ray. (e, f) P1 at 3 years showing typical facial features of Nager syndrome. (g) Hypoplasia of right thumb in P1 (left thumb has been surgically removed). (h–j) Milder features of Nager syndrome in P2: asymmetric face, down-slanting palpebral fissures, malar hypoplasia, micrognathia and mild hypoplasia of the right thumb with no creases on its ventral surface.
The dysmorphologic examination revealed down-slanting palpebral fissures, sparse lower eyelashes, malar hypoplasia, severe micro-retrognathia, floating left thumb, type II hypoplasia of right thumb (according to Blauth classification) and limited forearm pronation–supination (Figure 1a). Speech delay was observed owing to the severely limited mandible movements. The clinical features suggested a diagnosis of Nager syndrome.
Audiometric and TC evaluations revealed a moderate bilateral conductive hearing loss with mild stenosis of the external auditory canals and bilateral dysplasia of the middle ear.
Milder features of Nager syndrome were also detected in both the proband’s mother and grandmother. The physical examination of the mother (P2) revealed asymmetric face, down-slanting palpebral fissures, malar hypoplasia, micro-retrognathia and type II hypoplasia of the right thumb (Figure 1h–j). The grandmother (P3) was born with unspecified mandible anomalies and cleft palate, which were both surgically corrected. The physical evaluation revealed asymmetric face, down-slanting palpebral fissures, mild micro-retrognathia and type II hypoplasia of the left thumb.
The propositus, his mother and grandmother have no siblings, and both great-grandparents are dead.
Genetic analyses
After obtaining written informed consent, genomic DNA was extracted from the proband’s peripheral leukocytes and from his parents and maternal grandmother’s saliva samples using the MagPurix Blood DNA Extraction Kit (Resnova, Rome, Italy) or the Oragene kit (Oragene DNA, DNA Genotek, Ottawa, ON, Canada).
SF3B4 mutation analysis was performed as previously reported.6 The coding sequences of other 24 genes associated with mandibulo-facial and/or ear malformations were isolated and captured using the HaloPlex Target Enrichment system (Agilent Technologies, Santa Clara, CA, USA; Supplementary Table S1); indexed DNA fragments libraries were generated according to the manufacturer’s protocol (version D.5) and sequenced on a MiSeq Dx instrument (Illumina, San Diego, CA, USA), with 150 bp paired-end sequencing. Variant calling and bioinformatic analyses for NGS experiments were performed using the SureCall software version v3.0.2.1 (Agilent Technologies). The reported Genbank accession numbers for human SF3B4 are NG_032777.1 and NM_005850.4. Exons are numbered as reported in NG_032777.1. Nucleotide sequences were aligned using the Multalin software version 5.4.1 (http://multalin.toulouse.inra.fr/multalin/). Putative splice site variants were analyzed in silico using: NNSPLICE version 0.9 (www.fruitfly.org/seq_tools/splice.html), Human Splicing Finder (www.umd.be/HSF/), NetGene2 v2.4 (http://www.cbs.dtu.dk/services/NetGene2/). The novel SF3B4 variant has been submitted to the LOVD database [www.lovd.nl/SF3B4 (patient ID 78575)].
Construction and expression of the minigene
A 857 bp fragment including the exon 3 of the SF3B4 gene and part of the upstream and downstream introns was amplified from the patient’s genomic DNA with specific primers carrying the XhoI and NotI restriction sites (F: 5′-CTTCTctcgagGGGTCCCAGCAGTATTCACA-3′ and R: 5′-CTTCTgcggccgccTGACTCAATGTCCCCTGTC-3′) to enable the cloning within β-globin intron 2 of the minigene vector previously generated in our laboratory (Figure 2c).11 One wild-type and one mutant clones were retained for expression analysis, which was performed in HEK293 cells as reported elsewhere.11 RT-PCR was carried out using primers specific for β-globin exon 2 and exon 3 to avoid the amplification of the endogenous transcript; single bands were excised from agarose gel and sequenced.

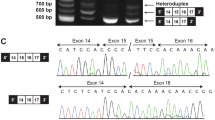
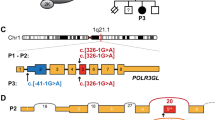
Genetic findings in our family with Nager syndrome. (a) SF3B4 sequencing shows the heterozygous c.417C>T substitution in P1, P2 and P3, and the wild-type allele in the unaffected proband’s father. (b) The c.417C>T variant affects a highly conserved nucleotide throughout evolution. (c) Schematic representation of the β-globin minigene construct used in this study. βEx1, βEx2, βEx3 refer to β-globin exon 1, 2 and 3. βInt1, βInt2 refer to β-globin intron 1 and 2. Wild-type and mutant exon 3 (SF3B4 Ex3) with at least 100 bp of the flanking introns (Int 2 and Int 3) were cloned into β-globin intron 2. (d) Total RNA extracted from cells transfected with the wild-type (WT), the mutant construct (MUT) and the empty vector (EV) was retro-transcribed and PCR-amplified using primers specific for β-globin exon 2 (βEX2F) and 3 (βEX3R) and separated by agarose gel electrophoresis. The mutant construct (MUT) showed an aberrant splicing compared with controls (WT) and yielded two bands. (e) Schematic representation of the structure of the transcripts obtained from the hybrid minigenes. The shorter transcript lacks an interstitial portion of exon 3 owing to the activation of a novel donor splice site (5′ SS) and the use of two different acceptor splice sites (3′ SS). The first and last nucleotides of the missing portion of exon 3 are indicated in blue and the novel SF3B4 pathogenic variant is highlighted in red. The upper band corresponds to the correctly spliced transcript.
Results and Discussion
The clinical picture of the proband (P1) was suggestive of Nager syndrome and thus we analyzed by direct sequencing the SF3B4 gene and identified a novel synonymous variant within exon 3 [c.417C>T p.(Gly139=)]. We also excluded by NGS technology disease-causing variants in a series of genes involved in craniofacial and/or ear malformations: all the variants identified in patient P1 were present in the available databases (minor allele frequencies are reported in Supplementary Table S2), except for the synonymous variant in SF3B4.
The following characteristics support the pathogenicity of the SF3B4 variant detected in our family: (i) it segregates with the pathologic phenotype, being present also in the mildly affected mother (P2) and maternal grandmother (P3; non-affected family members could not be tested since the propositus, his mother and grandmother do not have siblings; Figure 2a); (ii) it is a very rare variant which has never been reported in the literature nor detected in whole genome or exome sequencing projects (1000 genomes, ExAC and GO-ESP); (iii) it affects a highly conserved nucleotide throughout evolution (Figure 2b); and (iv) it is predicted to have deleterious effects on splicing by three different in silico tools, which suggested the activation of a cryptic splice site within SF3B4 exon 3 (Supplementary Table S3).12
As no patient’s RNA was available, to confirm its pathogenicity, we functionally characterized the synonymous variant using a β-globin hybrid minigene assay, which has been effectively used to validate potential splicing variants (Figure 2c).13 RT-PCR analysis from cells transfected with the mutant construct showed an aberrant splicing compared with controls and yielded two bands (Figure 2d): sequencing of the lower fragment revealed that it corresponds to shorter transcripts owing to the activation of a novel canonical AG/GT 5′ donor splice site within SF3B4 exon 3 and the use of two different AG acceptor sites (the first in position c.536_537, the second three nucleotides downstream) (Figure 2e). The corresponding messenger RNAs (mRNAs) result in the loss of 122 or 125 nucleotides, with the consequent formation of prematurely truncated proteins in both the cases (p.Gly139Glufs*6 or p.Asn140Leufs*4). Sequencing of the fainter upper band showed that it corresponds to the correctly spliced transcript, proving that the activity of the natural 5′ donor site of exon 3 is not completely abolished. These findings confirmed that the c.417C>T synonymous variant impairs normal pre-mRNA splicing and allows the partial production of the wild-type transcript, behaving like a hypomorphic allele.
Nager syndrome is a very rare acrofacial dysostosis. Until now, pathogenic heterozygous variants were detected in only 7 out of 12 multiplex families with a clinical diagnosis of Nager syndrome.4, 5 As previously observed, the affected individuals in our family showed an extremely variable phenotype, with a prenatally diagnosed, severe micrognatia in the proband, associated with marked preaxial malformations of the upper limbs. Conversely, the diagnosis of Nager syndrome had not been suspected in the mother nor in the grandmother until the birth of the proband.
The reasons of this variability are still unknown, but it likely relies on the interaction of genetic background and environmental factors. In addition, there is no evident genotype–phenotype correlations in Nager syndrome, with both frameshift and splicing pathogenic variants being reported in familial cases with patients showing variable clinical expressivity.4, 5 Our report further supports this notion and also suggests that even hypomorphic variants may be associated to both severe and mild forms of Nager syndrome. One possible explanation of the wide spectrum of severity of the syndrome in the three family members may be the different ratios between aberrant and wild-type constructs in critical tissues during the early embryo development. In any case, the lack of clear genotype–phenotype correlations makes genetic counseling particularly difficult for families with this syndrome.
In conclusion, our work underlines that it is crucial not to underestimate the possible effects on splicing of synonymous variants, especially when their frequency is compatible with the prevalence of the disease and no other variants are detected and confirmed the efficacy of minigene assays to provide functional data on the consequences of variants on transcript processing in the absence of the patient’s RNA.
References
Chemke J, Mogilner BM, Ben-Itzhak I, Zurkowski L, Ophir D : Autosomal recessive inheritance of Nager acrofacial dysostosis. J Med Genet 1988; 25: 230–232.
Hall BD : Nager acrofacial dysostosis: autosomal dominant inheritance in mild to moderately affected mother and lethally affected phocomelic son. Am J Med Genet 1989; 33: 394–397.
Aylsworth AS, Lin AE, Friedman PA : Nager acrofacial dysostosis: male-to-male transmission in 2 families. Am J Med Genet 1991; 41: 83–88.
Bernier FP, Caluseriu O, Ng S et al :Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet 2012; 90: 925–933.
Petit F, Escande F, Jourdain AS et al :Nager syndrome: confirmation of SF3B4 haploinsufficiency as the major cause. Clin Genet 2014; 86: 246–251.
Czeschik JC, Voigt C, Alanay Y et al :Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet 2013; 132: 885–898.
Castori M, Bottillo I, D’Angelantonio D et al :A 22-week-old fetus with Nager syndrome and congenital diaphragmatic hernia due to a novel SF3B4 mutation. Mol Syndromol 2014; 5: 241–244.
McPherson E, Zaleski C, Ye Z, Lin S : Rodriguez syndrome with SF3B4 mutation: a severe form of Nager syndrome? Am J Med Genet A 2014; 164A: 1841–1845.
Lund IC, Vestergaard EM, Christensen R, Uldbjerg N, Becher N : Prenatal diagnosis of Nager syndrome in a 12-week-old fetus with a whole gene deletion of SF3B4 by chromosomal microarray. Eur J Med Genet 2016; 59: 48–51.
Waggoner DJ, Ciske DJ, Dowton SB, Watson MS : Deletion of 1q in a patient with acrofacial dysostosis. Am J Med Genet 1999; 82: 301–304.
Forzan M, Salviati L, Pertegato V et al :Is CFTR 621+3A>G a cystic fibrosis causing mutation? J Hum Genet 2010; 55: 23–26.
Giorgi G, Casarin A, Trevisson E et al :Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin Chem Lab Med 2015; 53: 1719–1723.
Sharma N, Sosnay PR, Ramalho AS et al :Experimental assessment of splicing variants using expression minigenes and comparison with in silico predictions. Hum Mutat 2014; 35: 1249–1259.
Acknowledgements
This work was supported by Italian Ministry of Health [GR-2009-1578914 to ET], by University of Padova [CPDA140508/14 to ET] and by a grant from Fondazione Cariparo [to LS].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Cassina, M., Cerqua, C., Rossi, S. et al. A synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome. Eur J Hum Genet 25, 371–375 (2017). https://doi.org/10.1038/ejhg.2016.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.176
