
Abstract
MBD5-associated neurodevelopmental disorder (MAND) is an umbrella term that describes a group of disorders, 2q23.1 deletion syndrome, 2q23.1 duplication syndrome, and MBD5 variants, that affect the function of methyl-binding domain 5 (MBD5) and share a common set of neurodevelopmental, cognitive, and behavioral impairments. This review provides a comprehensive clinical and molecular synopsis of 2q23.1 deletion syndrome. Approaches to diagnosis, genetic counseling, and up-to-date management are summarized, followed by a discussion of the molecular and functional role of MBD5. Finally, we also include a brief summary of MBD5 variants that affect function of MBD5 and 2q23.1 duplication syndrome.
Similar content being viewed by others


In Brief
-
MBD5-associated neurodevelopmental disorder (MAND) includes deletion, duplication, and mutation of MBD5 (MRD1; OMIM 156200).
-
2q23.1 deletion syndrome is a dominant disorder characterized by intellectual disability, motor delay, severe speech impairment, seizures, and autism spectrum disorder.
-
The behavioral phenotype includes sleep disturbance, stereotypic repetitive behaviors, and short attention span.
-
2q23.1 deletion syndrome is caused by haploinsufficiency of MBD5 resulting from a deletion of the 2q23.1 chromosomal region in which MBD5 is located.
-
Individuals with pathogenic variants in MBD5 have a similar but typically milder 2q23.1 deletion syndrome-like phenotype.
-
Individuals with 2q23.1 duplication that include MBD5 have a phenotype similar to individuals with 2q23.1 deletion.
-
Management is symptomatic: treatment of seizures, sleep disturbance, and behaviors through pharmacological, diet, or other intervention strategies.
-
MBD5 is a dosage-sensitive gene.
-
MBD5 functions in transcriptional regulation.
-
Common pathways are shared between MBD5 and other syndromic ASD-associated genes.
Introduction
In the peak of genome-wide screening of individuals with intellectual disability (ID) using array comparative genomic hybridization (array-CGH), numerous novel chromosomal imbalances were identified. This genotype-first approach allowed for the identification of 2q23.1 deletions in patients with ‘pseudo-Angelman’ phenotypes1 and 2q23.1 duplications in patients with ASD-like phenotypes. Altered expression of MBD5 is the underlying etiology of these two disorders. Haploinsufficiency of MBD5 is responsible for the majority of the phenotypes present in 2q23.1 deletion syndrome, whereas overexpression of MBD5 leads to features of 2q23.1 duplication syndrome. Shadowing the arrival of array-CGH, the advent of genome-wide high-throughput sequencing of ID and ASD cohorts led to the identification of patients with pathogenic variants in MBD5.2 Phenotypes among patients with pathogenic variants, deletions, or duplications of MBD5 were noticeably similar,3 and accordingly, an umbrella term was coined, MBD5-associated neurodevelopmental disorder (MAND), to describe a group of disorders that have copy number variations methyl-binding domain 5 gene (MBD5) or sequence variants that affect function of MBD5 and share a common set of clinical features. Over the years, there have been significant studies on the clinical phenotypes of 2q23.1 deletion syndrome, yet the clinical phenotype for 2q23.1 duplication syndrome and MBD5 variant cases have yet to be thoroughly delineated. Further, the pathogenic consequences of haploinsufficiency of MBD5 have been extensively studied. Thus, the following review will depict our current understanding of the clinical and molecular aspects of 2q23.1 deletion syndrome. Finally, we discuss approach to diagnosis, genetic counseling, up-to-date management, and the role of MBD5 in 2q23.1 deletion syndrome.
Clinical overview of 2q23.1 deletion syndrome
2q23.1 deletion syndrome is a neurodevelopmental disorder that comprises ID, motor delay, severe speech impairment, seizures, sleep disturbances, autistic behaviors, and microcephaly with subtle minor dysmorphic features.1, 2, 4, 5 See Table 1 for the most prevalent features of 2q23.1 deletion syndrome.
Developmental delays
Developmental delays in 2q23.1 deletion syndrome are evident in all patients within the first year of life and include motor (gross and fine), language (receptive and expressive), and social skills. Motor delays with poor coordination and broad-based/ataxic gait are seen in over 70% of individuals,2, 5, 6 with the average age of walking 2–3 years of age.2, 6 Language development is severely impaired in individuals with 2q23.1 deletion syndrome. Most reported pediatric cases either lack speech entirely2 or have single words (2–3 word phrases),5, 7 and a few individuals speak 2–6 sentences.5, 6 Because of the limited speech and the broad-based ataxic gait, Angelman syndrome (AS) (MIM #105830) was considered in the diagnostic differential for some individuals who were eventually diagnosed with 2q23.1 deletions.5
Seizures
Seizures occur in more than 80% of children with 2q23.1 deletion syndrome.2, 6 Onset of seizures usually starts around 2 years of age.1, 2, 4, 5, 6, 7, 8, 9, 10, 11 Seizures have not been well characterized in this population; however, most individuals exhibit only one seizure type, of which absence spells, generalized tonic-clonic, atonic, sleep-related, and startle-induced atonic seizures have been described.1, 2, 4, 5, 6, 7, 8, 9, 10 A recent genomic copy number v ariant analysis implicated 2q23.1 deletions in Chinese children with infantile spasms.6 For the few cases reporting imaging studies, EEG patterns were non-specific, though a few patients were reported to have focal spikes and spike-wave complexes.2, 5, 6
Sleep disturbances
Individuals with 2q23.1 deletion have sleep disturbances that are similar to those observed in Smith–Magenis syndrome (SMS) (MIM# 182290), AS, and fragile X syndrome (FXS) (MIM# 300624), supporting the approach to the diagnostic differential for many of the initial cases reported.2, 5 Sleep disturbances are present in about 80% of the children with 2q23.1 deletion syndrome6 and include waking 6–8 times per night, apparent night terrors in the early part of sleep, and waking in the early hours of the morning.1, 2, 3, 4, 5, 6, 7, 8, 10 In a recent study of the characterization of the sleep problems in the 2q23.1 deletion syndrome population,12 a substantial portion of the children with 2q23.1 deletion did not sleep well at night and thus, had excessive daytime sleepiness likely due to poor and inconsistent sleep.12 Parents used interventions to ensure proper sleep for their children, including regular naps, containment (enclosed beds, locked doors) to prevent their child from wandering at night, and medications to promote sleep, such as melatonin.12
Other behaviors
The behavioral features of 2q23.1 deletion syndrome are very similar to other syndromic autism spectrum disorders. In infancy, hypotonia contributes to feeding difficulties for over 90% of the 2q23.1 population.2, 6 Short attention span and autistic-like behaviors, including gaze avoidance, inattention, and repetitive behaviors, are reported in more than 80% of individuals.13 Stereotypic and repetitive behaviors consisting of repetitive hand movements, teeth grinding, and the chewing of hands and are prevalent in >80% of the 2q23.1 deletion patients.2, 6 The stereotypic hand movements in many of these patients led to the initial suspicion of Rett syndrome (RTT) (MIM# 312750).5 Self-injury and aggression are seen in greater than 60% of the 2q23.1 deletion patients,2, 6 which, when coupled with sleep disturbance, led clinicians to consider SMS in the initial diagnostic assessment.2 Other behaviors mentioned in ~9% of reported patients are anxiety, hyperactivity, inappropriate happy demeanor, and social withdrawal.2, 6
Craniofacial and skeletal anomalies and other features
The majority of individuals with 2q23.1 deletion syndrome have mild craniofacial defects, but the anomalies are not uniform across the population (Figure 1). Reported facial features include broad forehead, structural nasal abnormalities (short nose and depressed or broad nasal bridge), outer ear abnormalities (simple protruding ear lobes), thick/arched eyebrows, downturned mouth corners, everted lower lip, tented upper lip, and thin upper lip.2, 6 Skeletal abnormalities are also not consistently seen in persons with 2q23.1 deletion syndrome. Although reports of hand and foot anomalies exist, the type of anomalies are wide ranging and not uniform and include small hands and feet (~75.0%), fifth finger clinodactyly (~70%), generalized brachydactyly (40.9%), short fifth digit of hands and feet (40%), and sandal gap (~33%).2, 6 Other skeletal abnormalities that are mentioned in at least two patients each are hip dysplasia, joint laxity, and scoliosis.2, 6

Craniofacial features of 2q23.1 deletion syndrome. Individuals with 2q23.1 deletion syndrome exhibit a broad forehead, bulbous nasal tip, thin upper lip, open mouth, and simple protruding ear lobes. (a) 12-month-old female. (b) 1 year 7-month-old male. (c) 2 year 2-month-old male. (d) 20-year-old male.
A variety of other minor anomalies have been reported, including constipation in >80% of the 2q23.1 deletion population.2, 6 About 11% of children with 2q23.1 deletion have cardiovascular abnormalities that consist of atrial septal defect, ventricular septal defect, and pulmonary stenosis.2, 6
Molecular and genetic basis of 2q23.1 deletion syndrome
2q23.1 deletion region
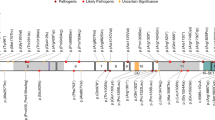
Typically in de novo deletion syndromes, haploinsufficiency of genes within the smallest region of overlap across deletion cases is considered causative of the phenotype.10 In the earliest published studies of 2q23.1 deletions, the common deletion region was found to involve two genes, MBD5 and EPC2 (see Figure 2, blue boxes).1, 2, 6, 14 One of these cases, 29195, showed a 200-kb deletion in 2q23.1 of only the MBD5 gene (see Figure 2, red box), which led to postulations that MBD5 could be the causative gene for this syndrome.15 A larger study by Talkowski et al.2 supported this hypothesis when the analysis of different-sized 2q23.1 deletions of 65 subjects revealed deletions of 2q23.1 ranging from small deletions of 38 kb to >19 Mb and two translocations within MBD5. Interestingly, there were no common deletion breakpoints among all 2q23.1 deletions evaluated in this or other studies, suggesting that 2q23.1 deletions most likely do not occur through low-copy-repeat-mediated nonallelic homologous recombination, as do deletion syndromes that show recurrent deletions.16, 17 It has been suggested that 2q23.1 deletions could be deletion mediated by recently delineated genomic rearrangement mechanism Fork Stalling and Template Switching (FoSTeS)/microhomology-mediated break-induced replication (MMBIR).9

Delineation of the critical region for 2q23.1 deletion syndrome. Schematic representation of MBD5 containing deletions previously reported in the literature. Boxes represent the minimum size of the deletions and the horizontal lines extend through gaps in coverage to show the maximum deletion sizes. Single asterisks indicate cases known to be de novo. Blue boxes represent the earlier published cases1,8 of 2q23.1 deletion syndrome before the study by Talkowski et al.2 Red box represents the first reported 2q23.1 deletion case that had only MBD5 deleted.15 Green boxes represent MBD5-specific deletions published in Talkowski et al.2 The two vertical black lines show the location of MBD5 and the smallest region of overlap between the deletions.
Upon aligning the 65 deletion regions, the study found that the smallest region of overlap (SRO) was defined to one gene, MBD5.2 No other genes were included in the SRO or were present in deletions that did not also involve MBD5. Patients presented with various deletions of MBD5, including 5′-noncoding exons (Figure 2, green boxes), coding exon deletion (Figure 2, green boxes), and large 2q23.1 deletions involving other genes (Figure 2).2 In addition, MBD5 mRNA expression levels for these various deletions were significantly reduced (22.5–55.4%; P<0.0001), further supporting haploinsufficiency of MBD5 as the causative mechanism behind the syndrome.2
Structure of the MBD5 gene
MBD5 has two known isoforms, 1 (UniProtKB ID # Q9P267-1) and 2 (UniProtKB ID # Q9P267-2).18 Isoform 1 is the longer primary isoform and has 1448 amino acids encoded by exons 6–15 (Figure 3a).18 The protein has two conserved domains, a MBD and a proline-tryptophan-tryptophan-proline (PWWP) domain, both of which may be found in chromatin-associated proteins (Figure 3b). Isoform 2 is encoded by exons 6–9, with intron 9 being retained (Figure 3c), resulting in a protein of 851 amino acids that does not contain the PWWP domain (Figure 3d). Isoforms 1 and 2 share amino acids 1–841 in common.18 Expression studies showed that isoform 1 is expressed in all tissues but highly expressed in brain and testis, while isoform 2 is expressed in all tissues but highly expressed in brain and ovaries.18

MBD5 gene and protein isoforms. (a) Coding exons of human MBD5 (exons 6–15) are shown in red. Translation of these exons yields protein isoform 1. (b) Protein isoform 1 is composed of a conserved MBD, a proline-rich segment (P-rich), a PWWP domain, and putative nuclear localization signals (NLS). (c) Coding exons of MBD5 (6–9) are shown in red. Translation of these exons yields protein isoform 2. (d) Protein isoform 2 is composed of a conserved MBD domain, a proline-rich segment (P-rich), and putative NLS.
MBD5 variants
MBD5-specific variants have been identified in a cohort of autism cases that also exhibit the phenotype of 2q23.1 deletion syndrome, including ID, severe speech impairment, seizures, and autistic-like behavioral problems (see Supplementary Table 1 for list of published MBD5 variants likely to affect function, GenBank accession numbers: NM_018328.4, and NP_060798.2).2, 7, 15, 19, 20, 21, 22 Variants were submitted to http://www.ncbi.nlm.nih.gov/clinvar/?term=MBD5[gene]). The identification of mutations in MBD5 in individuals with significantly overlapping features verified that MBD5 was the critical gene responsible for the core features of 2q23.1 deletion syndrome. In a survey of MBD5 coding variations among 747 ASD subjects compared with 2043 non-ASD subjects analyzed by whole-exome sequencing, six ASD patients had inherited heterozygous missense variants in MBD5, c.236G>A p.(Gly79Glu) (P=0.012) and c.3143C>T p.(Thr1048Ile),2 that were absent in controls, supporting that MBD5 variants contribute to ASD risk. In another comparable study of 287 patients with ASD, 32 changes were identified in MBD5 further supporting the significant role of MBD5 in the genetic etiology of autism.20 In a more recent study, Sanger sequencing of 78 subjects with a phenotype similar to 2q23.1 deletion syndrome found a nonsense variant, c.440C>G p.(Ser147*), in one patient.7 In the latest study, an analysis of 41 patients with moderate to severe ID identified a patient with an MBD5 variant, c.340_347del p.(Lys114Glyfs*35).19 Patients with MBD5 variants described here all exhibited developmental delay, motor delay, language impairment, behavioral impairment, and autistic-like behavioral problems, such as repetitive behaviors and social impairments, and seizures (Table 1). These features are present in all 2q23.1 deletion syndrome patients. However, patients with MBD5 variants have not yet been reported to exhibit any of the other frequent or occasional features prevalent in 2q23.1 deletion syndrome (see Table 1). This may be due to the fact that most mutation cases have been identified in larger cohorts where phenotypic characterization is less detailed. Whether the degree of severity observed in patients with MBD5 variants is comparable to that of 2q23.1 deletion cases still needs to be further investigated. Further, functional studies on some of these MBD5 variants (Supplementary Table) should occur to see if these variants affect the function of MBD5, and further clinical investigation is necessary to more fully understand the phenotypic spectrum associated with MBD5 variants.
Genotype–phenotype correlation of 2q23.1 deletion syndrome
Studies have shown that haploinsufficiency of MBD5 significantly impacts gene regulation in pathways associated with phenotypes typically observed in 2q23.1 deletions. In Talkowski et al.,2 ~84% of phenotypic features evaluated were consistently observed in both MBD5-specific deletion and 2q23.1 deletion cases, suggesting that the overall syndrome phenotype is primarily due to MBD5 haploinsufficiency.2 Developmental delay, motor delay, language impairment, seizures, behavioral problems, several craniofacial manifestations, and constipation were present in all or a majority of MBD5-specific disruptions and 2q23.1 deletions involving MBD5, supporting a significant role for MBD5 in these features.2
The core features of 2q23.1 deletion syndrome have been primarily attributed to MBD5 dosage. However, three genes (ORC4, KIF5C, and EPC2) just outside of the SRO could contribute to the phenotypic spectrum observed in 2q23.1 deletion subjects. Based on literature findings and predicted function, ORC4, KIF5C, and/or EPC2 could contribute to features such as microcephaly or other neurobehavioral phenotypes (Table 2 and Figure 2). Overall, while genes in the deletion region could modify and contribute to the variability of the phenotype, the core phenotype can be safely attributed to haploinsufficiency of MBD5.
Diagnosis and testing
Due to the significant similarity of phenotypic features observed in 2q23.1 deletion syndrome to other neurodevelopmental syndromes, such as AS, SMS, RTT, FXS, Kleefstra (MIM# 610253), and Pitt–Hopkins (MIM# 610954) syndromes, a clinical diagnosis or targeted testing based solely on phenotype is difficult. These disorders all share the core neurodevelopmental and behavioral characteristics of 2q23.1 deletion syndrome including ID, developmental delay, speech delay, motor delay, hypotonia, sleep disturbances, seizures, and autistic-like behavioral problems. An array-CGH confirms the diagnosis of 2q23.1 deletion syndrome in most cases (~90%). Thus, standard care that includes an array-CGH as an initial evaluation of developmental delays, will identify most 2q23.1 deletions. However, if this test proves negative, targeted gene sequencing with autism gene panels or whole-exome sequencing to identify MBD5 variants or other genes involved in pathways crucial to the phenotypes exhibited by the patient is the next step.
Genetic counseling and management
Genetic counseling
2q23.1 deletion syndrome is a dominant disorder caused by altered functional gene dosage of MBD5. Haploinsufficiency of MBD5 resulting from a deletion of the 2q23.1 chromosomal region in which MBD5 is located or a variant that affects function of MBD5 can lead to features listed in Table 1. Most individuals with 2q23.1 deletion syndrome have a de novo deleterious change in MBD5. However, in some cases, probands have inherited the variant from a parent in an autosomal dominant manner, suggesting incomplete penetrance and/or variable expressivity may be associated with this condition.2, 6 For the most accurate assessment of recurrence risk and proper counseling for the family, parental testing is recommended. If a parent carries the variant, the risk to offspring in any pregnancy is 50%; however, when parental karyotypes or MBD5 sequencing (as appropriate) are normal, the recurrence risk for future pregnancies is reduced to the general population risk. Gonadal mosaicism, while not yet reported for MBD5 deletions, is a possibility and should be communicated to the families.23 Although the theoretical risk for recurrence in sibs of a proband is very low, several multi-generation families exist.2, 6 Prenatal diagnosis through CMA for pregnancies at increased risk can be done if the genetic diagnosis has been established in a previously affected family member.
Predicting the severity associated with variants that exhibit reduced penetrance, whether a MBD5 variant or small 2q23.1 deletion, is difficult; however, these variants are associated with an increased risk for ASD.2, 7, 20, 24 Further, the 2q23.1 genomic region may have an important role in the predisposition for autism. Three studies, Talkowski et al.,2 Bonnet et al.,7 and Cukier et al.20 found MBD5 variants in children that were inherited from an apparently healthy parent.2, 7 Whether these inherited mutations contribute to the phenotype of the patient despite the inheritance from apparently healthy parent, requires future studies. Thus, it could be likely that while the proband shows evidence of ASD and the classic phenotypes of 2q23.1 deletion syndrome, the apparently healthy family members who have the same mutation could have a milder form of autism or it is possible that other genetic variants in the family are modifying the phenotypes. Thus, counseling the parent is highly necessary as well. However, more studies are required to more fully understand the phenotypic consequences of inherited MBD5 variants and to confirm variants are not rare polymorphisms that have no effect on protein function.
Management
A multidisciplinary approach is required to target the individual features and specific deficits identified in this syndrome. Care by specialists in the following fields is necessary: medical genetics, speech and language therapy, occupational and physical therapy, child development, neurology, nutrition/feeding, and ophthalmology. Infants would benefit from enrollment in an early intervention program. Routine management is necessary for seizures, language impairments, sleep disturbances, and constipation. School-age children will benefit from an individualized educational program. Speech therapy to include nonverbal methods of communication, including sign language and computer-assisted technologies, should be introduced early and developed as a way for communication given the poor or absent speech in most individuals with this disorder. Ongoing routine care and surveillance is recommended. Periodic reevaluation by a medical geneticist to provide new recommendations and information regarding 2q23.1 deletion syndrome should be scheduled. Further, periodic neurodevelopmental, developmental, and behavioral pediatric evaluation to assist in the management of cognitive and behavioral problems is recommended. Parent support groups such as the 2q23.1 MDS support group (https://www.facebook.com/2q23.1) or (www.2q23.org) offer tremendous practical day-to-day support to families.
Pharmacological and dietary approaches may help in alleviating co-morbidities associated with 2q23.1 deletion syndrome, including sleep disturbance, seizures, and behavioral problems. Many parents reported using daily pharmacologics such as melatonin, clonidine, and trazadone to cope with the sleep disturbances.12 Anticonvulsant medications such as valproate, clonazepam, zonisamide, and clobazam are used to treat the seizures and appear to be effective in reducing the incidence of seizures.1, 2, 4, 5, 6, 7, 8, 9, 10 Some 2q23.1 deletion syndrome case reports suggest that modified diets, such as gluten-free and dairy-free, could help with behavioral problems in children, and a dietary trial is an option to determine any positive response.2, 4, 5, 6 A lack of response to diet change would support a return to normal diet. In addition, some children are on a high-fat, low-carbohydrate ketogenic diet to moderate seizures; however, additional studies are required to fully inform this treatment recommendation,2, 5, 6 and this diet should be given under supervision of a registered dietitian and physician because this diet can lead to poor growth and poor weight gain. Overall, there are still not enough data to fully evaluate potential effects of diet on behavior or seizures.
Phenotype and genotype characterization of 2q23.1 duplication syndrome
MAND includes 2q23.1 duplication
The establishment of 2q23.1 deletions as a neurodevelopmental disorder led to investigation of duplications in this region. About 40 cases of 2q23.1 microduplications have been documented.3 The duplications range from 68 kb to 53.7 Mb, encompassing a region that includes MBD5.3 These duplications are nonrecurrent but mirror the regions impacted in 2q23.1 deletions. Upon alignment of the 2q23.1 duplications, the smallest region of overlap was defined to one gene, MBD5, common to all duplications. In addition, MBD5 mRNA expression levels for these various duplications were significantly elevated (1.5–1.7-fold increase; P<0.0001).3 However, it has not yet been determined whether small duplications disrupting part or all of MBD5 could lead to elevated expression levels. The phenotype of 2q23.1 duplication still needs to be refined; however, most individuals are indistinguishable from persons with 2q23.1 deletion syndrome. The clinical features that are shared between 2q23.1 duplication and 2q23.1 deletion patients are ID, developmental delay, motor delay, severe language impairment, infantile hypotonia, sleep disturbances, behavioral problems, and autistic-like features (Table 1). The following phenotypes present in 2q23.1 deletion cases: postnatal growth retardation, microcephaly, open mouth, small hands and feet, cardiovascular abnormalities, and constipation, were not noted in 2q23.1 duplication cases (Table 1).
Status of research in MAND
MBD5 is a dosage-sensitive gene
2q23.1 deletion syndrome and 2q23.1 duplication syndrome are associated with reciprocal deletions and duplications within chromosome region 2q23.1, with altered dosage of MBD5 leading to the core phenotypes (developmental delay, motor delay, language impairment, behavioral problems, and autistic-like symptoms) of both disorders (Figure 4).3 In Mullegama et al.,12 comparison of MBD5 pathogenic variants, and MBD5 deletions show that the core features of 2q23.1 deletion syndrome are seen in patients with MBD5 mutations and 2q23.1 duplication syndrome suggesting that MBD5 is a dosage sensitive gene.3 Thus, it was suggested that MBD5 and chromosome 2q23.1 should be added to an expanding list of dosage-sensitive genomic regions that result in reciprocal deletion and duplication of neurodevelopmental syndromes.3 Studies show that MBD5 mRNA expression levels in lymphocytes from individuals with 2q23.1 deletions ranged from approximately 0.22- to 0.59-fold of normal2 (Figure 4b). In addition, MBD5 mRNA expression levels were approximately 1.52–1.83-fold across all 2q23.1 duplications3 (Figure 4b). The phenotypes associated with a deletion or duplication of MBD5 combined with the gene expression changes observed support MBD5 as a dosage-sensitive gene, and thus, proper dosage of this gene is vital for normal neurodevelopment. Overall, a tight balance is required for MBD5 dosage.

MBD5 is a dosage-sensitive gene. (a) When chromosomal region 2q23.1 is altered due to either genomic imbalance or disruption due to translocation, manifestation of either 2q23.1 deletion syndrome or 2q23.1 duplication syndrome arises. (b) MBD5 mRNA expression analysis can assess the impact of a genomic abnormality on this dosage-sensitive gene. MBD5 duplication typically impacts mRNA expression, leading to increased levels ranging from 1.50- to 1.80-fold over normal levels, while an MBD5 deletion or disruption typically reduces mRNA expression to ~0.5-fold of normal (ranging from 0.65- to 0.20-fold). (c) The core phenotypes present in both deletion and duplication syndromes are similar and include ID, seizures (less prevalent in the duplication syndrome), developmental delays, and autism spectrum disorder.
Mbd5 mouse models recapitulate 2q23.1 deletion syndrome
Mouse models have been effective tools to understand the pathogenesis of 2q23.1 deletion syndrome and will be the key tools in future to aid in designing therapeutic strategies. Several different models alter or abolish Mbd5 expression in mice and include an Mbd5 knockout (Mbd5−/−),25 a brain-specific Mbd5 conditional knockout (Mbd5f/−, NestinCre),25 and a heterozygous hypomorph (Mbd5+/GT)26 (Table 3). The mouse models and more specifically, the Mbd5+/GT, nicely recapitulate aspects of the human condition25, 26 but also reveal findings that could help lead toward better understanding of the disorder and of MBD5 function. The Mbd5−/− mice exhibit early postnatal lethality,25 which necessitated the creation of a Mbd5 hypomorphic mouse, Mbd5+/GT, which was viable. The Mbd5+/GT mice exhibited behavioral abnormalities with neuronal function deficits, consistent with the 2q23.1 deletion syndrome human phenotype. Mbd5+/GT mice are smaller than WT, have an abnormal nasal bone development, and exhibit motor coordination impairment, which are present in children with 2q23.1 deletion syndrome. In addition, supporting the theory that MBD5 has a role in autism and ID, Mbd5+/GT mice exhibited abnormal social behaviors and clear indication of learning deficits and memory impairment (Table 3).26 Overall, more detailed phenotyping of these mice to determine if they exhibit seizures and circadian deficits would be beneficial. Further, Mbd5 transgenic mice overexpressing Mbd5 have yet to be described and would provide yet another model of the human condition.
MBD5 functions in transcriptional regulation
Prior to the identification of MBD5 as the causative gene for 2q23.1 deletion syndrome, little was known about MBD5. MBD5 belongs to the family of the methyl-CpG-binding domain (MBD) family of proteins, which includes MBD1-6 and MeCP2, the causative gene for Rett syndrome. The MBD family members have key roles in regulating gene transcription. Most members participate in chromatin remodeling and are known to mediate gene silencing through their MBD motifs.27 In an alignment of MBD family MBD regions, the MBD5 MBD has two major differences: a deletion of nine amino acids in the first third of the MBD and an insertion of six amino acids in the last third of the domain.18 Thus, a logical question to ask is whether or not MBD5 acts like MeCP2 and other MBD family members. Camarena et al.26 showed through immunocytochemistry experiments that MBD5 localizes to non-heterochromatin regions of the nucleus, which suggests that MBD5 acts as a transcriptional activator. Tao et al.27 suggested a connection between Mbd5 and the histone acetyltransferase, Kat2a, which is linked to memory formation and metabolism.27, 28 Kat2a is known to control glucose metabolism through transcriptional repression of Ppargcla,29 and Mbd5−/− mice have shown perturbed glucose homeostasis and heightened insulin sensitivity.27 In conclusion, further investigation to determine if MBD5 works directly/indirectly with KAT2A to mediate other physiological processes in addition to iron and glucose metabolism is required.
Common pathways shared between MBD5 and other syndromic ASD-associated genes
A growing theme in autism research purports that despite the genetic heterogeneity of ASDs, many associated genes converge in interconnected functional pathways.30 These interconnected pathways when disrupted result in the common phenotypes such as ID, sleep disturbance, language impairment, seizures, and behaviors present in many syndromic ASDs. Recently, 2q23.1 deletion syndrome, SMS (RAI1), and FXS (FMR1) were shown to share common sleep phenotypes.12 In addition, this study found that MBD5 and RAI1, along with FMR1, are involved in circadian and mTOR signaling pathways, both associated with sleep disturbance.12 MBD5 appears to share molecular relationships with ASD-associated genes, including protein-binding relationships with FMR1,31 APC,32 and MEF2C32 (Figure 5). Further, based on our co-expression studies, haploinsufficiency of MBD5 results in dysregulation of other ASD-associated genes, including UBE3A (AS), RAI1 (SMS), TCF4 (PTHS), MEF2C (OMIM#613443), and FMR1 (FXS) (Figure 5).33 Network database analyses utilizing databases such as GeneMANIA (www.genemania.org) and SFARI Gene (https://gene.sfari.org/) show molecular connections between MBD5 and several ASD associated genes (Figure 5). The dysregulation of any of these genes can impact pathways and thus, alter risks for phenotypes affecting sleep, language, and seizures that are present in 2q23.1 deletion patients. Whether MBD5 directly or indirectly regulates some of these genes remains to be determined. Identifying common neurobiological pathways will fundamentally advance our understanding toward treating the phenotypes seen in overlapping ASDs through knowledge-based therapeutic interventions.

MBD5 and ASD-associated genes. MBD5 has molecular interactions with ASD-associated genes, APC, FMR1, MEF2C, RAI1, TCF4, and UBE3A. These genes could be functioning together in pathways that affect phenotypic outcomes in 2q23.1 deletion syndrome and other ASDs. Green lines represent DNA interactions. Orange lines represent RNA binding interactions. Purple lines represent co-expression relationships. Red lines represent protein-binding. Figure was created from data from Gene Mania (http://www.genemania.org/) and SFARI Gene (https://gene.sfari.org/autdb/Welcome.do).
Conclusions
MAND encompasses a group of disorders associated with variation of MBD5 that may include chromosomal deletions, duplications, disruptions, or intragenic single nucleotide variation. Haploinsufficiency of MBD5 results in a complex syndrome of ID, with significant developmental delays, seizures, and autism spectrum disorder. The clinical presentation is not entirely distinct from other syndromic ID or ASDs. Consequently, diagnosis through clinical features can be difficult and requires consideration and analysis of high-resolution microarrays and/or targeted sequencing of MBD5. Further, the clinical features of 2q23.1 duplication syndrome and variants that affect functions of MBD5 are similar to 2q23.1 deletion syndrome, and more detailed phenotypic studies are needed to understand fully the phenotypes associated with 2q23.1 duplication and MBD5 pathogenic variants. MBD5 is required for proper neuronal function and thus, could likely be critical for processes such as learning, memory, and behavior. MBD5 acts as a transcriptional regulator, and proper regulation of MBD5 gene expression is likely important to the pathogenesis of ASD. The functional role of MBD5 is still not fully understood; thus, further studies are required. Strides are being made in understanding the molecular pathogenesis through mouse models that successfully recapitulate the clinical syndrome, as well as cell-based studies to examine molecular networks. Basic research on the molecular relationship between MBD5 and other ASD-associated genes is currently being done toward finding the pathways involved in common phenotypes such as sleep, language, epilepsy, and behaviors that could be critical for targeted therapeutic interventions.
Variant submission
Variants submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/?term=MBD5[gene]): SCV000264470, SCV000264471, SCV000264472, SCV000264473, SCV000264474, SCV000264475, SCV000264476, SCV000264477, SCV000264478, SCV000264479, and SCV000264480.
Change history
12 August 2015
This article has been amended since online publication. A corrigendum also appears in this issue.
References
Jaillard S, Dubourg C, Gerard-Blanluet M et al: 2q23.1 microdeletion identified by array comparative genomic hybridisation: an emerging phenotype with Angelman-like features? J Med Genet 2009; 46: 847–855.
Talkowski ME, Mullegama SV, Rosenfeld JA et al: Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 2011; 89: 551–563.
Mullegama SV, Rosenfeld JA, Orellana C et al: Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet 2014; 22: 57–63.
Noh GJ, Graham JM Jr : 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur J Med Genet 2012; 55: 59–62.
van Bon BW, Koolen DA, Brueton L et al: The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur J Hum Genet 2010; 18: 163–170.
Hodge JC, Mitchell E, Pillalamarri V et al: Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry 2014; 19: 368–379.
Bonnet C, Ali Khan A, Bresso E et al: Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur J Hum Genet 2013; 21: 1457–1461.
Williams SR, Mullegama SV, Rosenfeld JA et al: Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet 2010; 18: 436–441.
Motobayashi M, Nishimura-Tadaki A, Inaba Y et al: Neurodevelopmental features in 2q23.1 microdeletion syndrome: report of a new patient with intractable seizures and review of literature. Am J Med Genet A 2012; 158 A: 861–868.
Chung BH, Stavropoulos J, Marshall CR, Weksberg R, Scherer SW, Yoon G : 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am J Med Genet A 2011; 155 A: 424–429.
Shichiji M, Ito Y, Shimojima K et al: A cryptic microdeletion including MBD5 occurring within the breakpoint of a reciprocal translocation between chromosomes 2 and 5 in a patient with developmental delay and obesity. Am J Med Genet A 2013; 161 A: 850–855.
Mullegama SV, Pugliesi L, Burns B et al: MBD5 haploinsufficiency is associated with sleep disturbance and disrupts circadian pathways common to Smith-Magenis and fragile X syndromes. Eur J Hum Genet 2014; 23: 781–789.
Tan WH, Bird LM, Thibert RL, Williams CA : If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A 2014; 164 A: 975–992.
Burns B, Schmidt K, Williams SR, Kim S, Girirajan S, Elsea SH : Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum Mol Genet 2010; 19: 4026–4042.
Wagenstaller J, Spranger S, Lorenz-Depiereux B et al: Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet 2007; 81: 768–779.
El-Hattab AW, Zhang F, Maxim R et al: Deletion and duplication of 15q24: molecular mechanisms and potential modification by additional copy number variants. Genet Med 2010; 12: 573–586.
Boerkoel CF, Inoue K, Reiter LT, Warner LE, Lupski JR : Molecular mechanisms for CMT1A duplication and HNPP deletion. Ann N Y Acad Sci 1999; 883: 22–35.
Laget S, Joulie M, Le Masson F et al: The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS One 2010; 5: e11982.
Hamdan FF, Srour M, Capo-Chichi JM et al: De novo mutations in moderate or severe intellectual disability. PLoS Genet 2014; 10: e1004772.
Cukier HN, Lee JM, Ma D et al: The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res 2012; 5: 385–397.
Kleefstra T, Kramer JM, Neveling K et al: Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012; 91: 73–82.
O'Roak BJ, Vives L, Fu W et al: Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338: 1619–1622.
Campbell IM, Stewart JR, James RA et al: Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet 2014; 95: 345–359.
Noh GJ, Graham JM Jr : 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur J Med Genet 2012; 55: 354–357.
Du Y, Liu B, Guo F et al: The essential role of Mbd5 in the regulation of somatic growth and glucose homeostasis in mice. PLoS One 2012; 7: e47358.
Camarena V, Cao L, Abad C et al: Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome. EMBO Mol Med 2014; 6: 1003–1015.
Tao Y, Wu Q, Guo X, Zhang Z, Shen Y, Wang F : MBD5 regulates iron metabolism via methylation-independent genomic targeting of Fth1 through KAT2A in mice. Br J Haematol 2014; 166: 279–291.
Stilling RM, Ronicke R, Benito E et al: K-Lysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J 2014; 33: 1912–1927.
Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P : GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab 2006; 3: 429–438.
Ceroni F, Sagar A, Simpson NH et al: A deletion involving CD38 and BST1 results in a fusion transcript in a patient with autism and asthma. Autism Res 2014; 7: 254–263.
Darnell JC, Van Driesche SJ, Zhang C et al: FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011; 146: 247–261.
Bandyopadhyay S, Chiang CY, Srivastava J et al: A human MAP kinase interactome. Nat Methods 2010; 7: 801–805.
Mullegama SV, Alaimo JT, Chen L, Elsea SH : Phenotypic and molecular convergence of 2q23.1 deletion syndrome with other neurodevelopmental syndromes associated with autism spectrum disorder. Int J Mol Sci 2015; 16: 7627–7643.
Acknowledgements
We thank the Fondation Jérôme-Lejeune for funding a portion of the 2q23.1 deletion syndrome investigations. We thank Rachel D. Wills for help gathering papers and helping to create a table. We thank Drs Joseph T. Alaimo and Roberto Mendoza-Londono for critically reading the manuscript.
Author contributions
The contributions of the authors are as follows: SHE is the principal investigator of this project and contributed to the writing of the manuscript. SVM designed the outline, created the figures and tables, and wrote the manuscript. SVM and SHE critically revised the manuscript, and all authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Mullegama, S., Elsea, S. Clinical and Molecular Aspects of MBD5-Associated Neurodevelopmental Disorder (MAND). Eur J Hum Genet 24, 1235–1243 (2016). https://doi.org/10.1038/ejhg.2016.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.35
This article is cited by
-
Oligogenic inheritance in severe adult obesity
International Journal of Obesity (2024)
-
Five years of experience in the Epigenetics and Chromatin Clinic: what have we learned and where do we go from here?
Human Genetics (2023)
-
Exploring the genetic etiology of drug-resistant epilepsy: incorporation of exome sequencing into practice
Acta Neurologica Belgica (2022)
-
Emerging multifaceted roles of BAP1 complexes in biological processes
Cell Death Discovery (2021)
-
Composite Sleep Problems Observed Across Smith–Magenis Syndrome, MBD5-Associated Neurodevelopmental Disorder, Pitt–Hopkins Syndrome, and ASD
Journal of Autism and Developmental Disorders (2021)
