
Abstract
De novo GNAO1 variants have been found in four patients including three patients with Ohtahara syndrome and one patient with childhood epilepsy. In addition, two patients showed involuntary movements, suggesting that GNAO1 variants can cause various neurological phenotypes. Here we report an additional four patients with de novo missense GNAO1 variants, one of which was identical to that of the previously reported. All the three novel variants were predicted to impair Gαo function by structural evaluation. Two patients showed early-onset epileptic encephalopathy, presenting with migrating or multifocal partial seizures in their clinical course, but the remaining two patients showed no or a few seizures. All the four patients showed severe intellectual disability, motor developmental delay, and involuntary movements. Progressive cerebral atrophy and thin corpus callosum were common features in brain images. Our study demonstrated that GNAO1 variants can cause involuntary movements and severe developmental delay with/without seizures, including various types of early-onset epileptic encephalopathy.
Similar content being viewed by others


Introduction
Early-onset epileptic encephalopathies (EOEEs) are a group of neurological disorders characterized by impairments of development and intractable seizures from early infancy,1 including Ohtahara syndrome (OS) and migrating partial seizures of infancy (MPSI).2, 3 OS is the most severe and the earliest form of epileptic encephalopathy,4 and MPSI is characterized by migrating polymorphous focal seizures starting within the first 6 months.3
GNAO1 (MIM 139311) encodes an α-subunit of heterotrimeric guanine nucleotide-binding proteins (Gαo), which is extremely abundant in the brain tissue.5 We previously reported de novo GNAO1 variants in four patients with epileptic encephalopathy.6 Three of them showed OS and one patient had childhood epilepsy. Interestingly, two of them showed involuntary movements, suggesting that GNAO1 variants can cause various neurological phenotypes with variable types of seizures.
In this study, we report an additional four patients with de novo missense GNAO1 variants, expanding the phenotypic spectrum of GNAO1 variants.
Materials and methods
DNA samples and subjects
A total of 234 patients with infantile epilepsy were newly recruited as a second cohort. In addition, three patients with intellectual disability and involuntary movements were included in this study. Clinical information and peripheral blood samples were obtained after written informed consent was given. DNA was extracted using standard methods. Experimental protocols were approved by the institutional review board of Yokohama City University School of Medicine.
Whole-exome sequencing (WES)
Genomic DNA was captured using the SureSelect Human All Exon v4 Kit (51 Mb; Agilent Technologies, Santa Clara, CA, USA) and sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Exome data processing, variant calling, and variant annotation were performed as previously described.7 Variants were checked by using the database of our 575 in-house control exomes. In four patients with GNAO1 variants, trio-based WES was performed for one patient (patient 3), whereas the other three were identified by WES of proband only. All the GNAO1 variants detected by WES were confirmed by Sanger sequencing using trio samples (patients and their parents). De novo GNAO1 variants were deposited in a gene-specific database (http://databases.lovd.nl/shared/genes/GNAO1).
Structural analysis
Free-energy change of the Gα-containing complexes upon the variant was calculated using the FoldX software (version 3.0β6).8 Each variant was introduced into the crystal structure of the Gα subunit in different complexed states: GDP-bound Gαiβγ heterotrimer (PDB code 1GG2),9 the nucleotide-free Gαsβγ heterotrimer in complex with an agonist-occupied monomeric β2 adrenergic receptor (β2AR) (PDB code 3SN6),10 the transition-state GTP analog (GDP+AlF4-)-bound Gαq in complex with its effector PLCβ (PDB code 3OHM),11 and the GTP analog (GTPγS)-bound Gαs in complex with the catalytic domains of adenylyl cyclase (AC) (PDB code 1AZS).12 In this calculation, ligand molecules including the guanine nucleotides were ignored.
Results
Genetic analysis
By WES, we identified 4 unrelated patients with GNAO1 variants (2 of 234 patients with infantile epilepsy and 2 of 3 patients with intellectual disability and involuntary movements) (Figure 1a). Sanger sequencing using patients’ and their parental samples confirmed that all the variants occurred de novo. The c.607G>A is a recurrent variant, which was previously found in a patient showing childhood epilepsy and involuntary movement.6 One variant (c.736G>A) specifically affects GNAO1 transcript variant 1, whereas the other three variants affect both transcript variants 1 and 2 (GenBank accession number NM_020988.2 and NM_138736.2, respectively). Web-based prediction tools suggested that these four variants could affect protein function (Supplementary Table 1). None of the four variants was found in the 6500 National Heart, Lung, and Blood Institute exomes or among our 575 in-house control exomes.

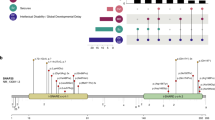
De novo GNAO1 variants in four patients. (a) Schematic representation of GNAO1 containing two transcript variants: variant 1 (GenBank accession number, NM_020988.2) and transcript variant 2 (NM_138736.2). The UTRs and coding regions are shown in white and dark blue rectangles, respectively. Previously reported three variants except for c.607G>A (red), which is recurrently identified in this study, are shown on the upper side. Five of seven variants occurred in common exons of two transcript variants. The c.736G>A and c.836 T>A substitutions affect specifically transcript variant 1 (exon 7; NM_020988.2). All the four variants in this study caused substitution at evolutionarily highly conserved amino acids. Homologous sequences were aligned using the CLUSTALW website. (b) Mapping of the variant sites on the crystal structures of Gα-containing complexes: the GDP-bound Gαiβγ heterotrimer (PDB code 1GG2),9 the nucleotide-free Gαsβγ heterotrimer in complex with an agonist-occupied monomeric β2AR (PDB code 3SN6),10 the transition-state GTP analog (GDP+AlF4−)-bound Gαq in complex with its effector PLCβ (PDB code 3OHM),11 and the GTP analog (GTPγS)-bound Gαs in complex with the catalytic domains of AC (PDB code 1AZS),12 from left to right, respectively. Gα, β, and γ subunits are colored in green, yellow, and pink, respectively, and the switch I and II regions in the Gα subunit are in cyan. The β2AR, PLCβ, and AC molecules are colored in gray, slate, and light brown, respectively. Guanine nucleotides are depicted as orange sticks. The variant sites are shown in red with their amino-acid number corresponding to human Gαo1 and, in parentheses, rat Gαi1 (UniProtKB/Swiss-Prot P10824), bovine Gαs (UniProtKB/Swiss-Prot P04896), mouse Gαq (UniProtKB/Swiss-Prot P21279), or bovine Gαs (UniProtKB/Swiss-Prot P04896). Molecular structures except for guanine nucleotides are shown as the space-filling representation from PyMOL (www.pymol.org). The illustrations below each model show a part of the G-protein activation process. (c) Free-energy change upon the amino-acid substitutions in each complex, calculated by the FoldX software.8
Structural consideration of variant effects.
To evaluate impacts of the found variants on Gα activity at the atomic structural level, we mapped the variant sites onto the crystal structures of some Gα-containing complexes in some different states: GDP-bound inactive, monomeric β2AR/G-protein coupling, and GTP-bound active states. In these complexes, variant effects are calculated as free-energy changes relative to wild type using the FoldX software.8 Arg209 is located within the switch II, a region important for guanine nucleotide-dependent regulation of downstream effectors such as PLCβ and AC (Figure 1b). In the GTP-bound active complexes (Figure 1b, right), the side chain of Arg209 makes a salt bridge with that of Glu246 (Supplementary Figure 1). Hence, the R209C and E246K variants should disrupt their interaction, resulting in destabilization of the Gα-containing complexes mainly in GTP-bound active states, which is consistent with the results of the FoldX calculation (Figure 1c).
Ala227 is close to the guanine nucleotide-binding site (Figure 1b) and makes a van der Waals contact with the methylene part of Lys271 located in the highly conserved NKXD motif, which is known to have an essential role in guanine ring binding (Supplementary Figure 1).13 Thus, the A227V variant is likely to affect the guanine nucleotide binding, although a relatively small impact of the variant on the protein structure was anticipated by the FoldX calculation (Figure 1c).
Clinical features of four patients with GNAO1 variants
Clinical features of four patients with GNAO1 variants are summarized in Table 1, and their case reports are shown in supplementary case reports. All four patients are female. No history of miscarriage was observed in these families. Two patients (patient 1 and 2) showed seizures during early-infantile period, and complex partial seizures accompanied by migrating or multifocal epileptiform discharges were observed in their clinical course (Figure 2a, Supplementary Figures 2 and 3). Their seizures are refractory to anti-epileptic drugs. In contrast, patients 3 and 4 showed no and few seizures, respectively, and both showed involuntary movements: athethosis in patient 3 and chorea in patient 4 (Table 1). Patients 1 and 2 also showed hand stereotypies and severe chorea, respectively. Severe intellectual disability and motor developmental delay are common in all the four patients. Brain magnetic resonance imaging (MRI) showed progressive cerebral atrophy and thin corpus callosum in three and two patients, respectively (Figures 2b–g). These data suggest that GNAO1 variants can cause both epileptic encephalopathy and involuntary movements accompanied by brain atrophy, but seizures are not always accompanied in some cases.

EEG and brain MRI features of patients with GNAO1 variants. (a) A monopolar recording of an ictal EEG of patient 2 at day 39 demonstrated the presence of sharp waves that initially emerged from the fronto-central region of the right hemisphere (left box), migrated into the contralateral side, and then evolved as an ictal pattern over the left hemisphere (right box). The scale bar at the bottom shows the duration (1 s) and the amplitude (50 μV). T1-weighted (b and g) and T2-weighted (c and d) axial images through the basal ganglia, and T1-weighted sagittal images (e, f). Cerebral atrophy in patient 1 (b), cerebral atrophy with delayed myelination in patient 2 (c), and almost normal findings in patient 3 (d) were observed. (e–g) Progressive cerebral and cerebellar atrophy, brainstem atrophy, and thin corpus callosum were observed in patient 4. m, months; y, years.
Discussion
Here we report four patients with de novo heterozygous missense variants in GNAO1. Patients with GNAO1 variants showed severe intellectual disability, motor developmental delay, and brain atrophy, further demonstrating that the aberration of Gαo affects normal brain development. In our previous report,6 three of four patients showed OS, which is characterized by tonic spasms mainly in the neonatal period.4 In this study, two patients showed migrating or multifocal complex partial seizures in their clinical course, similar to the findings of MPSI. Furthermore, involuntary movements such as chorea, athetosis, and hand stereotypies were observed in all the four patients, and two of them showed no or few seizures. Therefore, our report suggests that GNAO1 variants can cause wide clinical spectrum ranging from various types of EOEEs to involuntary movements without seizures. Together with our previous report,6 involuntary movements were observed in six of eight patients, indicating that involuntary movements may be one of the key features suspecting GNAO1 variants.
There are two patients harboring missense variants that specifically affected transcript variant 1, encoding Gαo1. Gαo1 is the most abundant Gαo protein in the mammalian brain.14, 15 Of the two patients, both showed severe intellectual disability and motor developmental delay, indicating that the affected Gαo1 function could cause neurological impairments. Although one of the two patients (patient 3 in this study) showed no seizures, another showed intractable seizures from early neonatal period. Further studies are needed for delineating possible differences in clinical features caused by variants affecting both transcripts and variants specifically affecting transcript variant 1.
It is surprising to note that all the eight patients reported to date are females, although GNAO1 is located on chromosome 16 (not X-linked). Gαo-deficient mice (Gnao1−/−) showed occasional seizures and generalized tremor with postnatal death,15, 16 and gain-of-function knock-in mutant mice (Gnao1G184S/+) also showed rare seizures, postnatal death, and a markedly increased frequency of interictal epileptiform discharges,17, 18 supporting that seizures and involuntary movement are the two major characteristic features caused by GNAO1 variants. However, no sex differences have been reported in these mouse models, and postnatal death was not influenced by sex in Gnao1G184S/+ mice.18 Therefore, the reason for sex bias in humans is currently unknown. Although more individuals with GNAO1 variants are necessary to make a conclusion on the female bias, it might be possible that males harboring GNAO1 variants could suffer from prenatal lethality.
Given the wide clinical spectrum of GNAO1 variants, it is possible that there is another allele modifying phenotypes. In this regard, it is interesting to note that lethality of Gnao1G184S/+ mutant mice was greatly influenced by strain differences: relatively normal life span with 100% survival in 129 background, whereas almost half of the mutants died by 40 weeks in B6 background.18 Genetic analysis revealed that the 129 allele in the Chr17: 41–70 Mb region has a protective effect from spontaneous death,18 suggesting that the phenotypes caused by GNAO1 variants are also modified by at least another locus in humans.
In conclusion, de novo GNAO1 variants were found in four female patients, two of whom showed involuntary movements but with few seizures. Genetic testing for GNAO1 should be considered in patients with EOEE or involuntary movement with severe developmental delay.
Accession codes
References
Mastrangelo M, Leuzzi V : Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr Neurol 2012; 46: 24–31.
Berg AT, Berkovic SF, Brodie MJ et al: Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010; 51: 676–685.
Coppola G : Malignant migrating partial seizures in infancy: an epilepsy syndrome of unknown etiology. Epilepsia 2009; 50: 49–51.
Ohtahara S, Yamatogi Y : Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 2006; 70: S58–S67.
Huff RM, Axton JM, Neer EJ : Physical and immunological characterization of a guanine nucleotide-binding protein purified from bovine cerebral cortex. J Biol Chem 1985; 260: 10864–10871.
Nakamura K, Kodera H, Akita T et al: De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013; 93: 496–505.
Saitsu H, Nishimura T, Muramatsu K et al: De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–449.
Guerois R, Nielsen JE, Serrano L : Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 2002; 320: 369–387.
Wall MA, Coleman DE, Lee E et al: The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell 1995; 83: 1047–1058.
Rasmussen SG, Choi HJ, Rosenbaum DM et al: Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007; 450: 383–387.
Waldo GL, Ricks TK, Hicks SN et al: Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 2010; 330: 974–980.
Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR : Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 1997; 278: 1907–1916.
Codina J, Birnbaumer L : Requirement for intramolecular domain interaction in activation of G protein alpha subunit by aluminum fluoride and GDP but not by GTP gamma S. J Biol Chem 1994; 269: 29339–29342.
Spicher K, Nuernberg B, Jager B, Rosenthal W, Schultz G : Heterogeneity of three electrophoretically distinct Go alpha-subunits in mammalian brain. FEBS Lett 1992; 307: 215–218.
Jiang M, Gold MS, Boulay G et al: Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA 1998; 95: 3269–3274.
Valenzuela D, Han X, Mende U et al: G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc Natl Acad Sci USA 1997; 94: 1727–1732.
Goldenstein BL, Nelson BW, Xu K et al: Regulator of G protein signaling protein suppression of Galphao protein-mediated alpha2A adrenergic receptor inhibition of mouse hippocampal CA3 epileptiform activity. Mol Pharmacol 2009; 75: 1222–1230.
Kehrl JM, Sahaya K, Dalton HM et al: Gain-of-function mutation in Gnao1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm Genome 2014; 25: 202–210.
Acknowledgements
We thank the patients and their families for their participation in this study. We also thank Nobuko Watanabe for her excellent technical assistance. This work was supported by a Grant-in-Aid for the Ministry of Health, Labour and Welfare of Japan; Grants-in-Aid for Scientific Research (B) (25293085, 25293235) and (A) (13313587); and challenging Exploratory Research (26670505) from the Japan Society for the Promotion of Science; the Takeda Science Foundation; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; the Strategic Research Program for Brain Sciences (11105137); and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12024421).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Saitsu, H., Fukai, R., Ben-Zeev, B. et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet 24, 129–134 (2016). https://doi.org/10.1038/ejhg.2015.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.92
This article is cited by
-
Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: from a severe G203R to a milder C215Y mutation
Acta Neuropathologica Communications (2022)
-
Genome-wide association study of stimulant dependence
Translational Psychiatry (2021)
-
Spectrum of movement disorders in GNAO1 encephalopathy: in-depth phenotyping and case-by-case analysis
Orphanet Journal of Rare Diseases (2020)
-
A meta-analysis of two high-risk prospective cohort studies reveals autism-specific transcriptional changes to chromatin, autoimmune, and environmental response genes in umbilical cord blood
Molecular Autism (2019)
-
GNAO1 mutation presenting as dyskinetic cerebral palsy
Neurological Sciences (2019)
