
Abstract
The difficulties arising from association analysis with rare variants underline the importance of suitable reference population cohorts, which integrate detailed spatial information. We analyzed a sample of 1684 individuals from Western France, who were genotyped at genome-wide level, from two cohorts D.E.S.I.R and CavsGen. We found that fine-scale population structure occurs at the scale of Western France, with distinct admixture proportions for individuals originating from the Brittany Region and the Vendée Department. Genetic differentiation increases with distance at a high rate in these two parts of Northwestern France and linkage disequilibrium is higher in Brittany suggesting a lower effective population size. When looking for genomic regions informative about Breton origin, we found two prominent associated regions that include the lactase region and the HLA complex. For both the lactase and the HLA regions, there is a low differentiation between Bretons and Irish, and this is also found at the genome-wide level. At a more refined scale, and within the Pays de la Loire Region, we also found evidence of fine-scale population structure, although principal component analysis showed that individuals from different departments cannot be confidently discriminated. Because of the evidence for fine-scale genetic structure in Western France, we anticipate that rare and geographically localized variants will be identified in future full-sequence analyses.
Similar content being viewed by others


Introduction
Advances in genotyping technologies have facilitated genome-wide association studies and have contributed to the identification of thousands of SNPs associated with disease1. However, most of the associated SNPs explain a modest proportion of heritability for most common diseases,2 and they do not provide variants directly applicable for diagnosis, prevention and treatment.3 With the advent of whole-genome sequencing, there is now the possibility to evaluate the role of genetic variants that were too rare to be picked up by GWAS.4 Because rare and potentially deleterious variants arose more recently than common variants, they were not yet eliminated by selection and they might provide most of the medically actionable alleles.3, 5 These alleles, because of their recent origin, tend to cluster within a lineage and therefore, many disease-related traits have been found in population isolates.6 Likewise, non-isolated populations displaying fine-scale genetic structure, within a short geographical distance, may be appropriate for gene mapping because they likely present high frequency of rare alleles.7
Moreover, there are specific difficulties arising from analyses based on rare variants, as they exhibit stronger stratification than common variants, which is not accounted for by existing statistical methods.8 The difficulties arising in rare-variant analyses underscore the importance of suitable reference population cohorts, which integrate detailed spatial information.8 In our study, we contribute to this effort by investigating fine-scale genetic structure in Western France based on 1684 genotyped individuals, for whom we have detailed geographic information. We provide a descriptive analysis of the pattern of population structure and genetic variation in Western France with the objective of building a reference panel useful for future association studies.
Since the seminal work of Menozzi et al,9 evidence has accumulated to show that allele frequencies vary within Europe, generating population structure at the continental scale.10, 11, 12 Evidence of population structure accumulates at the scale of European countries13, 14, 15, 16, 17, 18 and even at a finer scale.19, 20 Fine-scale genetic structure can result from different processes including founder effect, endogamy, historical migration, cultural barriers to gene flow or selection processes.11, 21, 22
Our study focuses on genetic variation in Western France as defined by Historical Geography literature. Before the 19th century, Western France included the ancient regions of Brittany–Anjou–Poitou and of Maine–Basse–Normandie (actually consisting on the departments of Calvados, Côtes-d’Armor, Finistère, Ille-et-Vilaine, Loire-Atlantique, Maine-et-Loire, Manche, Mayenne, Morbihan, Orne, Sarthe, Deux-Sèvres, Vendée, Vienne).23, 24 Western France is a large peninsula positioned in the northwest of France and delimited by the English Channel to the north, the Celtic and Atlantic Sea to the west and the Bay of Biscay to the south; the whole region covers ∼90 000 km2 extending ∼470 km from west to east. This part of France is now subdivided into different administrative regions further subdivided into departments (Figure 1 and Supplementary Figure 1).

Ancestry coefficients displayed using the RGB color model. To visualize the admixture coefficient, each individual is colored using a RGB (red–green–blue) coding scheme. The RGB coefficients are proportional to the ancestry coefficients obtained with admixture. Departments of the Pays de la Loire region (bottom left). Admixture coefficients for the different regions of WF. Following the same RGB coding scheme, this figures shows the variation of admixture coefficients in WF (bottom right).
We ascertained fine-scale population structure in Western France. Using principal component analysis (PCA) and model-based clustering, we investigated the relationship between genetic structure and place of birth. To better characterize fine-scale population structure, we merged individuals from Western France with the European Population Reference Sample (POPRES),25 and we computed measures of genetic proximity between Western France individuals and individuals from neighboring countries. We also better characterized fine-scale genetic variation by computing linkage disequilibrium (LD) and a local rate of genetic differentiation. Finally, we performed genome scans to map genomic regions correlated with population structure and to evaluate the genomic inflation of test statistics when not accounting for the place of birth.
Materials and methods
Samples
Analyses were performed with individuals from Western France coming from two different studies. The first study is the D.E.S.I.R. cohort, used to study the insulin resistance syndrome;26, 27 individuals from this cohort were born between 1930 and 1965 (median=1950). The second group consists of patients admitted to Nantes, Angers and Rennes hospitals, between 2004 and 2010, and included in the cohort CavsGen, for the study of calcific aortic valve stenosis, a common pathology affecting mainly individuals over 65 years old. Individuals from CavsGen were born between 1916 and 1949 (median=1931).
Individuals from both the D.E.S.I.R. and CavsGen cohorts were born in France and they came mainly from the Region Pays de la Loire (mainly departments of Loire-Atlantique, Maine-et-Loire and Vendée), but surrounding regions are also represented in the data (see Supplementary Table 1). The geographical locations of individuals were defined according to the place of birth, declared at the moment of enrollment in the cohorts. After excluding individuals with missing place of birth, the study population included 1684 unrelated individuals: 897 from D.E.S.I.R. and 787 from CavsGen. A third subgroup of 807 unrelated individuals, from the D.E.S.I.R. population, was used to replicate the relationship between genetic and geographical structure. This data set is called DESIR-REP in the following.
Genotyping
Samples were genotyped using the Axiom Genome-Wide CEU-1 Array (Affymetrix, Santa Clara, CA, USA). After quality control, we identified a set of 377 566 SNPs that were shared between D.E.S.I.R. and CavsGen. We pruned SNPs to account for LD (r2<0.2), which provided a total of 78 261 SNPs used for all analyses. In the LD analysis and the association scan, all shared SNPs were used.
The additional sample DESIR-REP was genotyped using the HumanHap300 Genotyping BeadChip (Illumina, San Diego, CA, USA). The quality control procedure was similar and based on allele frequency and LD.
Model-based clustering and PCA
To investigate the population structure in Western France, we performed model-based clustering as implemented in admixture.28 Admixture analysis was performed using K=2 and K=3 clusters. Before running admixture, we removed outlier individuals that would otherwise have formed a single cluster (four individuals in Maine-et-Loire and two individuals in Orne). Spatial kriging was used to interpolate ancestry coefficients. We also investigated population structure using PCA as implemented in the software smartpca (Cambridge, MA, USA) from the EIGENSOFT package.29
Identity-by-state statistics, LD and local differentiation
Pairwise identity-by-state (IBS) statistics were computed using Plink.30 LD was estimated using the r2 statistic. LD was computed based on the 377 566 SNPs in the data, after removal of the SNPs with minor allele frequencies under 5%. To compensate for unequal sampling in the different departments of interest, we used bootstrapping. In each department, 18 individuals were randomly selected (18 is the minimum sample size that is reached in the Manche Department) to compute r2 values. The process was repeated five times to average over several outcomes, and to compensate for sampling effects. Finally, local genetic differentiation was computed based on the pairwise Fst matrix between departments. Estimated with a Bayesian Kriging method, local genetic differentiation provides an estimate of the Fst measure of differentiation between a population living in the center of a department and a putative neighboring population living 30 km away.31
Genomic regions informative about Breton origin
We analyzed the association between a binary variable indicating Breton origin, based on the place of birth and the genotype. We used Plink to compute a χ2 statistic for each SNP, and we accounted for population stratification with the genomic inflation factor.30 We then considered 50-SNP windows and for each window, we performed a test of enrichment of SNPs informative about Breton origin. The P-values corresponding to the test of enrichment were obtained with a binomial test by computing the proportion of P-values smaller than 10−4 among each window of 50 SNPs. Because the data included two different cohorts (D.E.S.I.R. and CavsGen), we also performed an additional analysis to account for a potential cohort effect. We performed two independent GWAS on each of the cohorts and we combined the resulting P-values using Stouffer’s method32 for meta-analysis (also called inverse normal method).
Results
Population structure
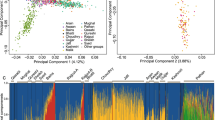
Performing a clustering analysis with admixture at both K=2 and K=3, we found that the admixture coefficients were correlated with geography (P<10−16 when regressing admixture coefficients with latitude and longitude). With K=2, admixture separated some of the Bretons from the rest of the sample (Supplementary Figure 2), and this cluster was again found with K=3 (green cluster in Figure 1). When averaging the ancestry of individuals within each department, we found that the ancestry component corresponding to the Bretons (green cluster) was larger in the three departments at the western end of Brittany (Finistère, Côtes d’Armor, Morbihan, see Supplementary Figure 3). With K=3, the ancestry of some individuals, many of them coming from the Vendée Department and from the south of the Maine-et-Loire department, differed from the rest of the sample (the blue cluster in Figure 1, see also Supplementary Figure 3).
The PCA confirmed the results obtained with admixture. The first principal component separated Bretons from the rest of the subjects (Figure 2 panel A) with individuals from the westernmost departments of Brittany having the lowest scores for PC1 (Supplementary Figure 4). The second component separated some individuals from the Vendée and Maine-et-Loire Departments from the rest of the sample (Figure 2 panel A and Supplementary Figure 4). The first two PC components were correlated with geography (P<10−16 when regressing the PC scores with latitude, longitude), also when accounting for genotyping plates and when considering the control DESIR-REP data set (P<10−9) (Supplementary Figure 5).

PCA of the CavsGen-DESIR data set. The envelopes correspond to the areas that contain 75% of the individuals of a given subdivision. (a) The envelopes correspond to the different regions of WF. Individuals from Brittany and from the Vendée and Maine-et-Loire Departments are displayed with larger crosses than the other individuals. (b) The envelopes correspond to the different departments of the Pays de la Loire Region, the Region with the majority of sampled individuals. Large crosses correspond to the barycentric coordinates of the individuals grouped by regions.
Then, we explored the PCA pattern for individuals born in the Pays de la Loire Region, because it was the most represented region in the data (926 out of 1684 sampled individuals). PCA showed genetic structure within the Pays de la Loire Region: some individuals from the western Loire-Atlantique department were located close to the Breton individuals, individuals from the northern Sarthe and Mayenne Departments were grouped with individuals from Normandy and some individuals from the southern Vendée Department and the eastern Maine-et-Loire department had the lowest scores for PC2 (Figure 2 panel B). However, PCA also showed considerable overlap between individuals from the different departments of the Region Pays de la Loire indicating that departmental origin could not be confidently assigned using the first two principal components.
Then, we merged the data with a collection of European individuals genotyped with the Affymetrix 500 K SNP panel (POPRES). From the first 10 PCs, we chose PC1 and PC3, because they were the most correlated with geography. Individuals from the DESIR-CavsGen data and from the control DESIR-REP data set were located between individuals from the UK, Ireland, Spain, the French-speaking part of Switzerland and the French POPRES population, which is consistent with their French origin (Supplementary Figures 6 and 7). Individuals from the DESIR-CavsGen data did not match exactly French individuals from POPRES, probably because of their westernmost origin.
When computing pairwise IBS, for individuals from this merged data set, we observed that the European countries showing the largest mean IBS with the DESIR-CavsGen individuals were France, UK, Ireland, followed by Germany and Belgium, followed by Spain and Italy (Figure 3 and Supplementary Figure 8). The IBS pattern differed according to French Regions, with the Brittany Region having a specific pattern. The countries that had the largest IBS with the Breton individuals were Ireland, UK and France in this order (Figure 3). The IBS between Irish and Bretons was significantly larger (P<10−12) than the IBS between Irish and individuals from the other regions of Western France (Normandy, Pays de Loire, Poitou-Charente, Centre), and this pattern was also found with the control DESIR-REP data set (Supplementary Figure 9). Conversely, the IBS between Spanish (or Italians) and Bretons was significantly smaller (P<10−12) than the IBS between Spanish (or Italian) and individuals from the other regions of Western France (Figure 3).

Mean IBS statistics between individuals from the French regions of the CavsGen-DESIR data set and individuals of POPRES from the neighboring countries of France.
Spatial variation of LD and population genetic differentiation
To study how LD varies in Western France, we computed LD within each Department. Figure 4 shows that the extent of LD is greater at the western end of Brittany and decreases when moving eastward. We also computed a statistic that measures the amount of local genetic differentiation for each department. Genetic differentiation increases with distance (the isolation-by-distance pattern, Supplementary Figure 10) at a rate that varies across Western France. The departments with the largest values of local genetic differentiation—measured in Fst per 30 km—are the three departments at the western end of Brittany as well as the Vendée department. In summary, both LD and local differentiation suggest that local effective population size is smaller in the western part of Brittany and local differentiation points to a specific pattern in the Vendée Department.

Spatial variation of local genetic differentiation (Fst at 30 km) and of LD (at 15 kb).
Most informative genomic regions about breton origin
To look for genomic regions that are informative about Breton origin, we performed genome scans by correlating SNPs to self-declared Breton origin. We found a genomic inflation factor of λ=1.26, confirming that there is a population structure in the data. When using Bonferonni correction, we found a SNP (rs6754311) in the lactase region significantly correlated with Breton origin. This SNP remained significantly correlated with Breton origin in a meta-analysis combining two independent association scans run for each cohort (CavsGen and D.E.S.I.R.). We also performed a window-based approach and searched for the 50-SNP regions enriched with ancestry-informative SNPs. The two genomic regions that were the most informative were around the lactase gene on chromosome 2, and in the HLA complex on chromosome 6 (Supplementary Figure 11). Around these two regions, we additionally computed for each 50-SNP window, the values of Fst between Ireland (POPRES) and the different French Regions. For both the HLA and the lactase regions, the Fst’s between Ireland and Brittany was markedly smaller than the Fst’s between Ireland and the other French Regions (Supplementary Figure 12).
For the lactase-persistence phenotype in Europe, the causative mutation is known and is located at SNP rs4988235. By computing the regional and departmental allele frequency of rs4988235, we found a west-to-east gradient of allele frequency with a frequency above 70% for 3 out of 4 departments in Brittany whereas the allele frequency decreased to between 45 and 55% in the easternmost departments of Western France (Supplementary Figure 13 and supplementary Table 2).
Discussion
There is currently ongoing interest in fine-scale population structure because it may impact association studies based on rare variants8 Our analysis shows that fine-scale population structure does occur at the scale of Western France. We found genetic ancestry related to Breton and Vendean origin. Uneven sampling can bias ascertainment of population structure33 but cannot cause the particular ancestry of Breton individuals because they are not overrepresented in the data set. Our results support the importance of isolation by distance in Western France because genetic differentiation between departments is well explained by geographical distances. The finding of fine-scale population structure in historical Western France is compatible with the historical records. Inner rural areas lacked long distance migration that resulted in isolated and static populations.34 Stability is also evident in the French rural population as a consequence of high endogamy and high percentages of marriages occurring within a short distance, at least until the 19th century.23 In his studies about marriage, R. Leprohon35 found that a high percentage of marriages took place between people living within a radius limited to 5–10 km. According to the analysis done by N. Pellen22 in Kerlouan, a Western French county, about 90% of mating was from the same village during the 17th and 18th centuries.
Patterns of genetic variation are partly generated by genetic drift defined as the random fluctuations of allele frequencies. When two populations diverge, genetic differentiation increases because of genetic drift. The smaller the effective population size, the larger the effect of genetic drift. We argue that the larger values of local Fst and of LD that were found in Brittany are explained by the enhanced effect of genetic drift in populations of lower effective population size.36 In the Vendée department, we posit that the presence of individuals with distinct ancestries (Figure 1) explains why only local Fst is increased. Because genetic differentiation increases more rapidly in these two regions, we expect that an excess of rare variants specific to these regions will be found with full-sequence data.
Because of population structure, association studies in Western France may generate false-positive associations. When using the Breton birthplace as an artificial phenotypic trait, two prominent associated genomic regions were found: the regions around the lactase gene and the HLA complex. By contrast to the lactase region, no single SNP within the HLA region was significantly associated with Breton origin. However, a correlation was found when looking for 50-SNP windows enriched with small enough P-values. The correlation between Breton origin and genotypes of the HLA region can be a true association, although there is strong LD within the HLA region,37 and this is a matter of concern when using window-based approaches. For these two regions informative about ancestry, there was a marked genetic proximity between Irish and Bretons, in terms of allele frequencies. The genetic proximity between Bretons and Irish was also found with Y chromosome haplogroups and on a genome-wide scale, possibly reflecting some degree of common Celtic origin.38, 39, 40 Both the lactase and the HLA regions are known to harbor prominent geographic variation in Europe and the increase in the lactase persistence allele in Brittany fits within the overall increase that is found when moving from southeastern to northwestern Europe.41 In particular, the frequency of the lactase persistent allele is high in Ireland (95%).41 The variation of lactase-persistence frequency is in line with the different admixture coefficients found for the Breton individuals pointing to something more than isolation-by-distance because of common ancestry between Bretons and individuals from the British isles. However, we acknowledge that admixture is not the only process that may have generated particular allele frequencies in Brittany; processes of positive selection that arose because of biological adaptation to agricultural practice can also explain spatial variation of gene frequencies.42
In summary, we emphasize the benefits of building reference panels with detailed information about geographic origin. In our study, we used geographic information to reveal fine-scale genetic structure in historical Western France. We found distinctive ancestries for Vendean and Breton individuals and the latter were found to have genetic proximities with Irish individuals. At a more subtle level, we also identified very fine-scale population structure within the Pays de la Loire Region. As for other European regions, such as the Netherlands, British Isles and Sardinia,18, 43, 44 it is important to build a comprehensive reference panel of Western France genetic variation with fine-grained geographic information in view of performing rare-variant association studies.
References
Hindorff LA, Sethupathy P, Junkins HA et al: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 2009; 106: 9362–9367.
Manolio TA, Collins FS, Cox NJ et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA : Clan genomics and the complex architecture of human disease. Cell 2011; 147: 32–43.
Cirulli ET, Goldstein DB : Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 2010; 11: 415–425.
McClellan J, King M-C : Genetic heterogeneity in human disease. Cell 2010; 141: 210–217.
Kristiansson K, Naukkarinen J, Peltonen L : Isolated populations and complex disease gene identification. Genome Biol 2008; 9: 109.
Henn BM, Gravel S, Moreno-Estrada A, Acevedo-Acevedo S, Bustamante CD : Fine-scale population structure and the era of next-generation sequencing. Hum Mol Genet 2010; 19: R221–R226.
Mathieson I, McVean G : Differential confounding of rare and common variants in spatially structured populations. Nat Genet 2012; 44: 243–246.
Menozzi P, Piazza A, Cavalli-Sforza L : Synthetic maps of human gene frequencies in Europeans. Science 1978; 201: 786–792.
Heath SC, Gut IG, Brennan P et al: Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet 2008; 16: 1413–1429.
Novembre J, Johnson T, Bryc K et al: Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Jay F, Sjödin P, Jakobsson M, Blum MGB : Anisotropic isolation by distance: the main orientations of human genetic differentiation. Mol Biol Evol 2013; 30: 513–525.
Humphreys K, Grankvist A, Leu M et al: The genetic structure of the Swedish population. PLoS One 2011; 6: e22547.
O’Dushlaine CT, Morris D, Moskvina V et al: Population structure and genome-wide patterns of variation in Ireland and Britain. Eur J Hum Genet 2010; 18: 1248–1254.
Salmela E, Lappalainen T, Liu J et al: Swedish population substructure revealed by genome-wide single nucleotide polymorphism data. PLoS One 2011; 6: e16747.
Di Gaetano C, Voglino F, Guarrera S et al: An overview of the genetic structure within the Italian population from genome-wide data. PLoS One 2012; 7: e43759.
Esko T, Mezzavilla M, Nelis M et al: Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur J Hum Genet 2012; 21: 659–665.
Abdellaoui A, Hottenga J-J, Knijff de P et al: Population structure, migration, and diversifying selection in the Netherlands. Eur J Hum Genet 2013; 21: 1277–1285.
O’Dushlaine C, McQuillan R, Weale ME et al: Genes predict village of origin in rural Europe. Eur J Hum Genet 2010; 18: 1269–1270.
Colonna V, Nutile T, Ferrucci RR et al: Comparing population structure as inferred from genealogical versus genetic information. Eur J Hum Genet 2009; 17: 1635–1641.
Biswas S, Scheinfeldt LB, Akey JM : Genome-wide insights into the patterns and determinants of fine-scale population structure in humans. Am J Hum Genet 2009; 84: 641–650.
Pellen N Hasard, coïncidence, prédestination... et s’il fallait plutôt regarder du côté de nos aïeux? Analyse démographique et historique des réseaux généalogiques et des structures familiales des patients atteints de mucoviscidose en Bretagne. Thèse de doctorat en démographie historique. Versailles, France: Université de Versailles-Saint-Quentin-en-Yvelines, 2012.
Blayo Y, Henry L : Données démographiques sur la Bretagne et l’Anjou de 1740 à 1829. Ann Démographie Hist 1967; 1967: 91–171.
Dupaquier J : La dynamique démographique de la France de l’Ouest au XIXe s. Norois 1988; 140: 409–414.
Nelson MR, Bryc K, King KS et al: The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet 2008; 83: 347–358.
Balkau B : [An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): epidemiologic data on the insulin resistance syndrome]. Rev Dépidémiologie Santé Publique 1996; 44: 373–375.
Sladek R, Rocheleau G, Rung J et al: A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445: 881–885.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Duforet-Frebourg N, Blum MGB : Non-stationary patterns of isolation-by-distance: inferring measures of local genetic differentiation with Bayesian kriging. Evolution 2013; 68: 1110–1123.
Stouffer SA, Suchman EA, Devinney LC, Star SA, Williams RM Jr : The American Soldier: Adjustment During Army Life. (Studies in Social Psychology in World War II, Volume 1). Princeton University Press, NJ, USA, 1949.
McVean G : A genealogical interpretation of principal components analysis. PLoS Genet 2009; 5: e1000686.
Hohenberg P : Migrations et fluctuations démographiques dans la France rurale, 1836-1901. Ann Économies Sociétés Civilis 1974; 29: 461–497.
Leprohon R : Vie et mort des Bretons sous Louis XIV. Brasparts, France: Bibliophiles de Bretagne, 1984.
Pimenoff VN, Laval G, Comas D et al: Similarity in recombination rate and linkage disequilibrium at CYP2C and CYP2D cytochrome P450 gene regions among Europeans indicates signs of selection and no advantage of using tagSNPs in population isolates. Pharmacogenet Genomics 2012; 22: 846–857.
Evseeva I, Nicodemus KK, Bonilla C, Tonks S, Bodmer WF : Linkage disequilibrium and age of HLA region SNPs in relation to classic HLA gene alleles within Europe. Eur J Hum Genet 2010; 18: 924–932.
McEvoy B, Richards M, Forster P, Bradley DG : The Longue Durée of genetic ancestry: multiple genetic marker systems and Celtic origins on the Atlantic facade of Europe. Am J Hum Genet 2004; 75: 693–702.
Dubut V, Chollet L, Murail P et al: mtDNA polymorphisms in five French groups: importance of regional sampling. Eur J Hum Genet 2004; 12: 293–300.
Busby GB, Brisighelli F, Sánchez-Diz P et al: The peopling of Europe and the cautionary tale of Y chromosome lineage R-M269. Proc Biol Sci 2012; 279: 884–892.
Itan Y, Jones BL, Ingram CJE, Swallow DM, Thomas MG : A worldwide correlation of lactase persistence phenotype and genotypes. BMC Evol Biol 2010; 10: 36.
Fix AG : Gene frequency clines in Europe: demic diffusion or natural selection? J R Anthropol Inst 1996; 2: 625–643.
Winney B, Boumertit A, Day T et al: People of the British Isles: preliminary analysis of genotypes and surnames in a UK-control population. Eur J Hum Genet 2012; 20: 203–210.
Chen W, Li B, Zeng Z et al: Genotype calling and haplotyping in parent-offspring trios. Genome Res 2013; 23: 142–151.
Acknowledgements
We thank Emmanuelle Bourcereau who was involved in patient recruitment of the CavsGen cohort. We also thank Professors Le Guerrier and Baufreton from University Hospitals of Rennes and Angers. We are grateful to the French Regional Council of Pays-de-la-Loire (Regional grant Biliv, grant VACARME - www.vacarme-project.org), the French Ministry of Health (PHRC/RNI-PROG/09/61: Génétique du rétrécissement aortique) and the French Ministry of Research (CavsGen project, ANR-13-BSV6-0011-01). We wish to thank the Fondation Genavie for its financial support. The POPRES data were obtained from dbGaP (accession no. phs000145.v3.p2). FS was funded by a Eurostars E!6490 Cardiomarks grant. MGBB is supported by the French National Research Agency (DATGEN project, ANR-2010-JCJC-1607-01). The D.E.S.I.R. study has been supported by INSERM contracts with CNAMTS, Lilly, Novartis Pharma and Sanofi-Aventis; by INSERM (Réseaux en Santé Publique, Interactions entre les déterminants de la santé), Cohortes Santé TGIR, the Association Diabète Risque Vasculaire, the Fédération Française de Cardiologie, La Fondation de France, ALFEDIAM, Société francophone du diabète, ONIVINS, Abbott, Ardix Medical, Bayer Diagnostics, Becton Dickinson, Cardionics, Merck Santé, Novo Nordisk, Pierre Fabre, Roche, Topcon.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The D.E.S.I.R. Study Group INSERM U1018: B Balkau, P Ducimetière, E Eschwège; INSERM U367: F Alhenc-Gelas; CHU D’Angers: A Girault; Bichat Hospital: F Fumeron, M Marre, R Roussel; CHU de Rennes: F. Bonnet; CNRS UMR8090, Lille: S Cauchi, P Froguel; Centres d’Examens de Santé: Alençon, Angers, Blois, Caen, Chateauroux, Chartres, Cholet, Le Mans, Orléans, Tours; Institute de Recherche Médecine Générale: J Cogneau; General practitioners of the Region; Institute inter-Regional pour la Santé: C Born, E Caces, M Cailleau, O Lantieri, JG Moreau, F Rakotozafy, J Tichet, S Vol.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Karakachoff, M., Duforet-Frebourg, N., Simonet, F. et al. Fine-scale human genetic structure in Western France. Eur J Hum Genet 23, 831–836 (2015). https://doi.org/10.1038/ejhg.2014.175
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.175
This article is cited by
-
The genetic structure of Norway
European Journal of Human Genetics (2021)
-
The genetic history of France
European Journal of Human Genetics (2020)
-
The Irish DNA Atlas: Revealing Fine-Scale Population Structure and History within Ireland
Scientific Reports (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}