
Abstract
The highly structured distribution of Y-chromosome haplogroups suggests that current patterns of variation may be informative of past population processes. However, limited phylogenetic resolution, particularly of subclades within haplogroup K, has obscured the relationships of lineages that are common across Eurasia. Here we genotype 13 new highly informative single-nucleotide polymorphisms in a worldwide sample of 4413 males that carry the derived allele at M526, and reconstruct an NRY haplogroup tree with significantly higher resolution for the major clade within haplogroup K, K-M526. Although K-M526 was previously characterized by a single polytomy of eight major branches, the phylogenetic structure of haplogroup K-M526 is now resolved into four major subclades (K2a–d). The largest of these subclades, K2b, is divided into two clusters: K2b1 and K2b2. K2b1 combines the previously known haplogroups M, S, K-P60 and K-P79, whereas K2b2 comprises haplogroups P and its subhaplogroups Q and R. Interestingly, the monophyletic group formed by haplogroups R and Q, which make up the majority of paternal lineages in Europe, Central Asia and the Americas, represents the only subclade with K2b that is not geographically restricted to Southeast Asia and Oceania. Estimates of the interval times for the branching events between M9 and P295 point to an initial rapid diversification process of K-M526 that likely occurred in Southeast Asia, with subsequent westward expansions of the ancestors of haplogroups R and Q.
Similar content being viewed by others
Introduction
One approach to deciphering the multilayered process of human dispersal has come from interpreting patterns of genetic variation at mtDNA and the non-recombining portion of the Y chromosome (NRY).1, 2, 3, 4, 5, 6, 7, 8 These haploid systems are subject to stronger genetic drift and large evolutionary variance, and may not render accurate signals of population processes by themselves. Yet, the considerable geographic structure at these loci suggests that current patterns of variation may be informative of past population processes. With the implicit assumption that groups dispersing in the Pleistocene were small and experienced strong and long-lasting bottlenecks, patterns of mtDNA and NRY variation have been deemed useful as starting points to formulate hypotheses about human demographic history.9
Human NRY variation is classified according to monophyletic groups of lineages (often referred to as haplogroups, clades or subclades) defined by single-nucleotide polymorphisms.10, 11 For example, the mutation M168 defines the lineage leading to a major clade (C–T) that contains almost all non-African Y chromosomes, and hence has been hypothesized to mark an early expansion of modern human populations out of Africa.1, 8, 10 Diversification of the clade defined by M168 resulted in multiple subclades, one of which is defined by the mutation M9 (haplogroup K) (Figure 1a) and contains multiple subclades that span Eurasia, Oceania and the Americas. It has been suggested that the M9 mutation arose somewhere in the Middle East shortly after anatomically modern humans dispersed from Africa.1

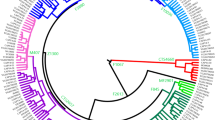
Phylogeny of haplogroup K before (a) and after (b) this study. In (a), haplogroups are named according to the nomenclature system in 2008,10 and in (b), haplogroups are renamed following the conventions of the YCC report.11 Mutations shown in italic-bold define new branches on the phylogenetic tree. The haplogroup M147 has not been assigned to the M526 haplogroup, but is represented here in gray.
Until now, the internal structure of the K haplogroup, especially the major subclade defined by the M9 mutation, has not been well characterized. The discovery of the M526 and P326 mutations1, 12 split this clade into two subclades, one containing chromosomes within the L and T haplogroups and the other containing several K lineages (K-M526*, K-M147, K-P60, K-P79, K-P261), as well as chromosomes within the M, NO, P and S haplogroups (Figure 1a). Although L and T haplogroups have relatively restricted distributions, K-M526 contains the majority of non-African Y chromosomes. The large number of broadly distributed subbranches of K-M526 has been suggested to reflect a rapid range expansion of K-M526 carriers soon after their origin somewhere in Central or East Asia, with subsequent local differentiation.1 However, the poorly resolved internal structure of this major haplogroup has limited inference on the geographic origin of its subhaplogroups. Here we map onto the NRY tree 13 mutations that greatly improve the phylogenetic resolution of haplogroup K-M526, and shed light on the geographic origin of haplogroup P.
Subjects and methods
We hierarchically genotyped 13 mutations in a sample of 4413 M526-derived Y chromosomes (see Supplementary Table S1 for a list of primers used for genotyping assays). This information was submitted to the International Society of Genetic Genealogy (http://www.isogg.org/tree/ISOGG_YDNA_SNP_Index.html). All samples were reported in previous publications.2, 13, 14, 15, 16, 17 These samples were collected according to procedures approved by the University of Arizona Human Subjects Committee. The relative lengths of periods between branching events along a lineage are estimated as described in Karafet et al.10 Briefly, the number of mutations in each of the disjoint periods follows a multinomial distribution, with parameters given by the ratio of the length of each period relative to the total time for the lineage, and the number of tries equal to the total number of mutations for the lineage. The fraction of time between two branching events can be estimated with confidence intervals using a binomial distribution.
Results
A set of 13 mutations was extracted either from sets of known polymorphisms, by comparisons that include low-coverage genome sequences,18 or discovered here by resequencing when genotyping previously known mutations (Supplementary Table S1). Six of these mutations define new K haplogroups containing previously undifferentiated K-M526 chromosomes (P315, P336, P378, P401, P402, and P403), three appear to be equivalent to previously described mutations (P304, P307 and P308) and four change the topology of haplogroup K by combining subhaplogroups (P331, P397, P399, and P405) (Figure 1b).
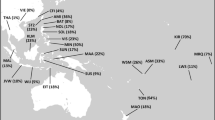
The P331 mutation is derived in almost all K-M526 chromosomes in our survey that are not in haplogroup NO, with the exception of chromosomes carrying the mutations P261 (observed in Bali), P402/403 (observed in Java) and a small number of K-M526* chromosomes, which we observe in the Indonesian islands of Sumatra and Sulawesi (Table 1). The rare haplogroup K-M147, originally reported in two individuals from South Asia,19 was not detected in our samples, and could not be placed accurately on the tree. All P331 chromosomes tested are derived either at both P397 and P399 (haplogroup K2b1) or at P295 (haplogroup P or K2b2). These two subclades display remarkably different geographic distributions (Table 1 and Figure 2). Haplogroup P-P295 (K2b2) with sister clades Q and R is by far the most frequent haplogroup in Europe, the Americas, and Central and South Asia. On the other hand, K2b1 is nearly restricted to eastern Indonesia, Oceania, Australia and the Philippines (Aeta), with very low incidence in Southeast Asia and western Indonesia (Table 1 and Figure 2).

Y-chromosome K-M526-derived haplogroups and their frequencies in a global sample of 7462 chromosomes. For haplogroup relationships see Figure 1.
Within K2b1 (K-P397) we report a new clade defined by the P405 mutation, which combines haplogroups K-P60, K-P79 and S-M230, as well as the newly defined K-P315 and K-P401 haplogoups (Figure 1b). Also contained within K2b1 are haplogroups M (M-P256) and the newly defined K-P336 and K-P378 haplogroups. Most of these newly described lineages are highly localized geographically to Island Southeast Asia/Oceania: K-P315 is confined to Melanesia (being observed in New Britain, Vanuatu and Papua New Guinea), K-P401 is detected in Vanuatu, K-P336 chromosomes are found primarily in Alor (but are also present in Borneo and Timor), K-P378 is found exclusively among the Aeta of the Philippines and K-P304 and K-P308 are observed only in Australia (and are equivalent to K-P60). Incidentally, the Y chromosome obtained from an ∼100-year-old lock of hair in Australia, initially identified as K-M526*,20 carries the derived allele at P304. The P295 mutation, previously assumed to be equivalent to 18 other mutations defining the haplogroup P,10 is derived in a broader group of chromosomes. In our worldwide sample of 7462 Y chromosomes, we observe the newly defined paragroup P-P295* in 83 chromosomes from Island Southeast Asia (Timor, Sumba, Sulawesi) and the Negrito Aeta population from Philippines (Table 1 and Figure 2).
We estimate the time between the most recent common ancestors of chromosomes carrying the M9 mutation and that of the subset of chromosomes carrying the P331 mutation using the probability distribution for mutations along a lineage as described in Karafet et al.10 We consider a set of 68 mutations that were uniformly ascertained from resequencing studies21 and that are distributed along the lineage connecting the common ancestor of chromosomes derived for M168 with the tip of a haplogroup R chromosome (see details in Karafet et al10). This set fails to indicate the presence of the K-M526 clade and of its subclade K-P331 (Figure 1), suggesting that K-M9 diversified rapidly after its first appearance. Considering that we do not observe any of the 68 mutations occurring between the common ancestor of K-M9 chromosomes and that of K-P331 chromosomes, we estimate that this interval of time was shorter than 4.3% of the TMRCA of M168 chromosomes (95% upper bound). Assuming ∼70 ky for the TMRCA of M168 chromosomes,10 we estimate the interval of time between the diversification of K-M9 and that of K-P331 to be <3 ky. This rapid diversification has also been assessed using whole Y-chromosome sequence data.22 In addition, we estimate the total time between the common ancestor of K-M9 and that of P-P295 to be <5 ky, and the time between the common ancestor P-P295 and that of P-P27 to be 12.3 ky (95% CI: 6.6–20 ky).
Discussion
In this study, we defined phylogenetic relationships of the sub-branches of K-M526 that were previously unknown. We observe a rapid diversification of the K-M526 subclade, which may be typical of a recovery following a bottleneck. However, a strict recovery may not be expected to result in patterns of genetic variation that are geographically structured. Populations experiencing rapid range expansion followed by low levels of gene flow may also see a rapid increase in the global number of lineages23 while having geographically structured patterns of genetic variation. A similar pattern of rapid diversification resulting in geographic structure of genetic variation has been observed for the more inclusive haplogroup F.22
The inference of the geographic location in which specific lineages diversified is often based upon the distributions of closely related haplogroups. For instance, if all lineages descending from a specific individual are found in the same geographic area, it is commonly inferred that the most recent common ancestor of those lineages also lived in that same geographic area. In some cases, gene flow may explain why not all derived lineages are present in the proposed area of diversification. For instance, the R-V88 haplogroup is almost completely restricted to Africa, yet closely related lineages in haplogroup R are extremely rare in sub-Saharan Africa.24, 25 Because the phylogenetic structure of haplogroup R is characterized by several consecutive basal splitting events leading to tip branches that are currently observed only outside Africa, it is extremely unlikely that haplogroup R diversified in Africa. Similarly, the phylogenetic structure of haplogroup K-M526 shows consecutive branching events (M526, P331 and P295), which appear to have rapidly diversified. With the exception of P-P27, all of the descendant lineages are located today in Southeast Asia and Oceania: K-M526*, K-P402, K-P261 and NO are the lineages most closely related to haplogroup K-P331, K-P397 is the sister lineage of P-P295 and the P-P295* lineages are the closest relatives of haplogroup P-P27 (Figure 1b). This pattern leads us to hypothesize a southeastern Asian origin for P-P295 and a later expansion of the ancestor of subhaplogroups R and Q into mainland Asia. An alternative explanation would involve an extinction event of ancestral P-P295* chromosomes everywhere in Asia. These scenarios are equally parsimonious. They involve either a migration event (P* chromosomes from Indonesia to mainland Asia) or an extinction event of P-P295* paragroup in Eurasia. However, given the geographic distribution of the P331 mutation, the immediate predecessor of P lineage and its likely origin in Southeast Asia/Indonesia, the existing evidence favors the first scenario. Interestingly, ancient DNA evidence suggests that haplogroup R1b – the current dominant lineage in western Europe – did not reach high frequencies until after the European Neolithic period as given in Lacan et al26, 27 and Pinhasi et al.28
In sum, our results support the hypothesis of a Southeast Asian/Oceanian center for the diversification of Oceanian K-haplogroup lineages and underscore the potential importance of Southeast Asia as a source of genetic variation for Eurasian populations. We propose that the patterns of Y-chromosome variation in the K haplogroup reflect a process of population fragmentation, likely associated with the early expansion of modern human populations into island Southeast Asia, and possibly also with rapidly changing sea levels,29 followed by a subsequent dispersal from the same area. While limited in their inferential power, our results warrant the exploration of a demographic model that includes a population expansion from island Southeast Asia into mainland Asia.
References
Chiaroni J, Underhill PA, Cavalli-Sforza LL : Y chromosome diversity, human expansion, drift, and cultural evolution. Proc Natl Acad Sci USA 2009; 106: 20174–20179.
Hammer MF, Karafet TM, Redd AJ et al: Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 2001; 18: 1189–1203.
Hudjashov G, Kivisild T, Underhill PA et al: Revealing the prehistoric settlement of Australia by Y chromosome and mtDNA analysis. Proc Natl Acad Sci USA 2007; 104: 8726–8730.
Hurles ME, Nicholson J, Bosch E, Renfrew C, Sykes BC, Jobling MA : Y chromosomal evidence for the origins of oceanic-speaking peoples. Genetics 2002; 160: 289–303.
Jobling MA, Tyler-Smith C : The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 2003; 4: 598–612.
Kayser M : The human genetic history of Oceania: near and remote views of dispersal. Curr Biol 2010; 20: R194–R201.
Macaulay V, Hill C, Achilli A et al: Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science 2005; 308: 1034–1036.
Underhill PA, Kivisild T : Use of Y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet 2007; 41: 539–564.
Barbujani G, Bertorelle G, Chikhi L : Evidence for Paleolithic and Neolithic gene flow in Europe. Am J Hum Genet 1998; 62: 488–492.
Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF : New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 2008; 18: 830–838.
YCC: A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res 2002; 12: 339–348.
Mendez FL, Karafet TM, Krahn T, Ostrer H, Soodyall H, Hammer MF : Increased resolution of Y chromosome haplogroup T defines relationships among populations of the Near East, Europe, and Africa. Hum Biol 2011; 83: 39–53.
Karafet T, Xu L, Du R et al: Paternal population history of East Asia: sources, patterns, and microevolutionary processes. Am J Hum Genet 2001; 69: 615–628.
Karafet TM, Hallmark B, Cox MP et al: Major east–west division underlies Y chromosome stratification across Indonesia. Mol Biol Evol 2010; 27: 1833–1844.
Karafet TM, Lansing JS, Redd AJ et al: Balinese Y-chromosome perspective on the peopling of Indonesia: genetic contributions from pre-neolithic hunter-gatherers, Austronesian farmers, and Indian traders. Hum Biol 2005; 77: 93–114.
Redd AJ, Roberts-Thomson J, Karafet T et al: Gene flow from the Indian subcontinent to Australia: evidence from the Y chromosome. Curr Biol 2002; 12: 673–677.
Tumonggor MK, Karafet TM, Karafet TM et al: The Indonesian archipelago: an ancient genetic highway linking Asia and the Pacific. J Hum Genet 2013; 58: 165–173.
Green RE, Krause J, Briggs AW et al: A draft sequence of the Neandertal genome. Science 2010; 328: 710–722.
Underhill PA, Shen P, Lin AA et al: Y chromosome sequence variation and the history of human populations. Nat Genet 2000; 26: 358–361.
Rasmussen M, Guo X, Wang Y et al: An Aboriginal Australian genome reveals separate human dispersals into Asia. Science 2011; 334: 94–98.
Hinds DA, Stuve LL, Nilsen GB et al: Whole-genome patterns of common DNA variation in three human populations. Science 2005; 307: 1072–1079.
Poznik GD, Henn BM, Yee MC et al: Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 2013; 341: 562–565.
Ray N, Currat M, Excoffier L : Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 2003; 20: 76–86.
Cruciani F, Santolamazza P, Shen P et al: A back migration from Asia to sub-Saharan Africa is supported by high-resolution analysis of human Y-chromosome haplotypes. Am J Hum Genet 2002; 70: 1197–1214.
Cruciani F, Trombetta B, Sellitto D et al: Human Y chromosome haplogroup R-V88: a paternal genetic record of early mid Holocene trans-Saharan connections and the spread of Chadic languages. Eur J Hum Genet 2010; 18: 800–807.
Lacan M, Keyser C, Ricaut FX et al: Ancient DNA reveals male diffusion through the Neolithic Mediterranean route. Proc Natl Acad Sci USA 2011; 108: 9788–9791.
Lacan M, Keyser C, Ricaut FX et al: Ancient DNA suggests the leading role played by men in the Neolithic dissemination. Proc Natl Acad Sci USA 2011; 108: 18255–18259.
Pinhasi R, Thomas MG, Hofreiter M, Currat M, Burger J : The genetic history of Europeans. Trends Genet 2012; 28: 496–505.
Pope KO, Terrell JE : Environmental setting of human migrations in the circum-Pacific region. J Biogeogr 2008; 35: 1–21.
Acknowledgements
We thank all the donors for providing DNA samples. This work was supported by the Salus Mundi Foundation to TMK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
Performed and designed experiments: TMK; analyzed the data: TMK, FLM, MFH; collected samples: HS, JSL; wrote the paper: TMK, FLM, MFH.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Karafet, T., Mendez, F., Sudoyo, H. et al. Improved phylogenetic resolution and rapid diversification of Y-chromosome haplogroup K-M526 in Southeast Asia. Eur J Hum Genet 23, 369–373 (2015). https://doi.org/10.1038/ejhg.2014.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.106
This article is cited by
-
Parallel signatures of Mycobacterium tuberculosis and human Y-chromosome phylogeography support the Two Layer model of East Asian population history
Communications Biology (2023)
-
Sequence analyses of Malaysian Indigenous communities reveal historical admixture between Hoabinhian hunter-gatherers and Neolithic farmers
Scientific Reports (2022)
-
The paternal and maternal genetic history of Vietnamese populations
European Journal of Human Genetics (2020)
-
Carriers of mitochondrial DNA macrohaplogroup L3 basal lineages migrated back to Africa from Asia around 70,000 years ago
BMC Evolutionary Biology (2018)
-
The study of human Y chromosome variation through ancient DNA
Human Genetics (2017)
