
Abstract
We present a study of five children from three unrelated Irish Traveller families presenting with primary ciliary dyskinesia (PCD). As previously characterized disorders in the Irish Traveller population are caused by common homozygous mutations, we hypothesised that all three PCD families shared the same recessive mutation. However, exome sequencing showed that there was no pathogenic homozygous mutation common to all families. This finding was supported by histology, which showed that each family has a different type of ciliary defect; transposition defect (family A), nude epithelium (family B) and absence of inner and outer dynein arms (family C). Therefore, each family was analysed independently using homozygosity mapping and exome sequencing. The affected siblings in family A share a novel 1 bp duplication in RSPH4A (NM_001161664.1:c.166dup; p.Arg56Profs*11), a radial-spoke head protein involved in ciliary movement. In family B, we identified three candidate genes (CCNO, KCNN3 and CDKN1C), with a 5-bp duplication in CCNO (NM_021147.3:c.258_262dup; p.Gln88Argfs*8) being the most likely cause of ciliary aplasia. This is the first study to implicate CCNO, a DNA repair gene reported to be involved in multiciliogenesis, in PCD. In family C, we identified a ∼3.5-kb deletion in DYX1C1, a neuronal migration gene previously associated with PCD. This is the first report of a disorder in the relatively small Irish Traveller population to be caused by >1 disease gene. Our study identified at least three different PCD genes in the Irish Traveller population, highlighting that one cannot always assume genetic homogeneity, even in small consanguineous populations.
Similar content being viewed by others

INTRODUCTION
Primary ciliary dyskinesia (PCD) (MIM#242650) is a heterogeneous autosomal recessive disorder characterised by impairment of mucociliary clearance. Clinically, PCD manifests as chronic bronchial sepsis and bronchiectasis, sinusitis and chronic secretory otitis media.1 In a large subset of PCD patients, laterality is randomised resulting in 50% of cases with abdominal and thoracic situs inversus. Male infertility and female sub-fertility is also common. Less common associations include oesophageal disease, biliary atresia, complex congenital heart disease and hydrocephalus.2 The estimated prevalence of PCD is 1:15 000–30 000 live births, although this is likely an underestimate as clinical under-diagnosis is common.1 The prevalence is increased in certain consanguineous or isolated populations and families.3
Mutations in genes that cause PCD result in defective cilia that move abnormally or are completely immotile. Ciliary ultrastructural abnormalities, such as defects of dynein arms, microtubules and connecting radial spokes, can be visualised by electron microscopy.4 PCD is associated with a high degree of genetic heterogeneity, with 28 disease genes identified to date. The type of ciliary defect in a patient is often an indication of the type of gene that is mutated in the individual (Table 1). Of the known causes of PCD, DNAH5 mutations are the most common and account for 15–22% of cases.5 However, the majority of PCD cases are still of unknown aetiology.
In this study, we report on five children from three unrelated consanguineous Irish Traveller families (families A–C) who presented with recurrent lower respiratory tract infections (LRTIs) (Figures 1a, 2a and 3a). A diagnosis of PCD was made on the basis of the combination of (1) presentation of classic clinical PCD features, (2) low nasal nitric oxide levels plus (3) ciliary abnormalities revealed by electron microscopy analysis of nasal ciliary brushings (Figures 1b–d and 3b and Supplementary Table S1) and (4) abnormal ciliary motility on video microscopy. As genetic disorders in the Traveller population are typically caused by a common homozygous mutation, we initially hypothesised that all five children shared the same recessive PCD mutation. We proposed to identify this shared mutation using whole-exome sequencing.

Irish Traveller family A. (a) Family A includes two siblings (II:1 and II:2) with PCD and glycogen storage disease (GSD) type III. Individual II:3 has GSD type III without PCD. DNA was available from the individuals denoted with a *. % GenHom; percentage of the autosomal genome located in a region of homozygosity >1 Mb in the affected children. (b–d) Transmission electron microscope analysis of bronchial epithelium samples from the Irish siblings with PCD. White arrows indicate cilia with normal ultrastructure. Black arrows indicate absence of the central pair. Dashed arrows indicate absence of the central pair and translocation of a peripheral pair to the center. (b) Ciliary cross-sections from child II:1 at × 20 000 magnification. Microvilli are evident. (c) Ciliary cross-sections from child II:2 at × 70 000 magnification. (d) A zoomed-in view of microtubule defects. (e) The RSPH4A NM_001161664.1:c.166dup duplication was validated by Sanger sequence analysis. The inverted triangle indicates the duplicated C base on the forward strand that causes a frameshift at residue 56 (Arg (R) to Pro (K)). The amino acid sequence is denoted using single letter database codes.

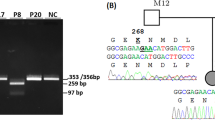
Irish Traveller family B. (a) Family B includes two affected siblings (IV:13 and IV:15) with PCD. TEM of nasal brushings from IV:13 showed nude epithelium, which may be a primary (ciliary aplasia) or secondary (infection) effect. DNA was available from the individuals denoted with a *. % GenHom; percentage of the autosomal genome located in a region of homozygosity >1 Mb in the affected children. (b) Sanger sequence validation of the 5 bp duplication (NM_021147.3:c.258_262dup) in exon 1 of CCNO. The GCCCG 5 bp duplication occurs immediately after two repeats of the same GCCCG sequence. The GCCCG sequence is repeated three times in succession in the patient (top), but only twice in the control sample (bottom). Each wild-type 5 bp sequence (repeat) is denoted by a dashed rectangle. The extra GCCCG sequence is marked with a red rectangle. The amino acid sequence is denoted using single letter database codes. The full colour version of this figure is available at European Journal of Human Genetics online.

Irish Traveller family C (a) A singleton (II:1) in family C was diagnosed with PCD. DNA was available from the individuals denoted with a *. % GenHom; percentage of the autosomal genome located in a region of homozygosity >1 Mb in the affected child. (b) Transmission electron microscope analysis of bronchial epithelium samples from patient C;II:1 typically showed absence of both inner and outer dynein arms. The left-hand panel shows a ciliary cross-section at × 50 000 magnification. The right-hand panel shows a zoomed-in view of the inner and outer dynein arm defects at × 80 000 magnification. (c) A homozygous ∼3.5-kb deletion, which includes exon 7 of DYX1C1, was identified by exome sequencing (II:1) and copy-number variant analysis in the affected child in family C. A screenshot of the Integrative Genomics Viewer shows a lack of sequence reads (pink/light blue boxes) across DYX1C1 exon 7 in patient II:1 compared with the control sample. DYX1C1 exons are shown as dark blue boxes at the bottom of the browser. The full colour version of this figure is available at European Journal of Human Genetics online.
MATERIALS AND METHODS
Clinical and diagnostic assessment
Written informed consent was obtained from patient guardians and the study protocol was approved by the ethics committee of Temple Street Children’s University Hospital (Ireland). A detailed medical history was taken from all individuals, and all were physically examined.
Family A (A;II:1 and A;II:2) initially presented to the respiratory service with a history of recurrent LRTI, chronic wet cough, persistent segmental collapse of the left lower lobe with likely bronchiectasis on CT thorax and documented hearing deficit on official testing. In addition to PCD, both children were diagnosed with glycogen storage disease (GSD) type III, early-onset severe cardiomyopathy and developmental delay (secondary to GSD III) and are closely monitored by both the metabolic, cardiology, ear nose and throat (ENT) and respiratory services. One of the two patients (A;II:1) also had a myelomeningocoele that was repaired at birth. A further sibling (A;II:3) has GSD type III without PCD. The two children with PCD and GSD type III (A;II:1 and A;II:2) are short in stature and both have PEG tubes in place for feeding.
Family B includes two siblings who presented with recurrent LRTIs. In addition, the elder sibling (B;IV:13) has a history of recurrent otitis media but the younger sibling (B;IV:15) does not. Neither have recurrent sinusitis or situs inversus. The younger sibling was identified early because of a high index of suspicion based on his elder brother’s diagnosis. Repeat nasal oxide screening tests were universally low at 30–50 p.p.b., indicative of PCD.
Child II:1 from family C presented with a neonatal pneumonia necessitating 16 days of intravenous antibiotics at birth. He also had significant left main stem bronchomalacia, identified at flexible bronchoscopy and bronchoalveolar lavage, resulting in recurrent left-sided pneumonias with rapid clinical deterioration.
Diagnostics and management
All patients regularly attend the ENT service for hearing and sinus monitoring and treatment. The older three patients have documented hearing loss (A;II:1, A;II:2 and B;IV:13) with child A;II:1 requiring a hearing aid within the last 2 years. Respiratory examination in the affected sib-pair from family A (II:1 and II:2) at baseline revealed reduced breath sounds at left bases, whereas the respiratory examination of the children from families B and C was normal. Progression of lung disease is monitored by repeated lung function testing (spirometry, total lung capacity (TLC) and diffusing capacity of the lung for carbon monoxide (DLCO)), chest radiographs and CT thorax where indicated. Children A;II:1 and A;II:2 have CT thorax evidence of segmental left-lobar collapse with bronchiectasis, the older sibling from family B (IV:13) has evidence of bronchical wall thickening but no bronchiectasis and the remaining two children (B;IV:15 and C;II:1) have no bronchiectasis on CT thorax.
Lung function testing using a body plethysmograph (Jaeger, Wurzburg, Germany) is reliable in children aged ∼≥5 years with good technique only. Owing to developmental delay in children II:1 and II:2 (family A), lung function technique has been poor in the past and results have been unreliable or unobtainable. More recently, child A;II:1 has mildly reduced spirometry, child A;II:2 has moderately reduced spirometry and the older patient in family B (IV:13) has moderately reduced spirometry with normal TLC and DLCO. The other two children (B;IV:15 and C;II:1) are too young to perform these tests reliably. Flexible bronchoscopy and bronchoalveolar lavage have been performed on three of the six children revealing significant copius mucus bilaterally in child A;II:1 and B;IV:13, and left main stem bronchomalacia with thick tenacious mucus in child C;II:1. All samples cultured haemophilus influenza. Further diagnostic assessment included detailed analysis of immune function (normal full blood count, IgG, IgM, IgA, IgE, IgG subclasses, tetanus diphtheria, haemophilus influenza and pneumococcal titres), a sweat test (normal) and out-ruling aspiration.
Children A;II:1, A;II:2, B;IV:13 and B;IV:15 are admitted to hospital every 3 months for prophylactic intravenous antibiotic therapy, nebulised rhDNase (pulmozyme, Genentech Inc, South San Fransisco, CA, USA) and chest physiotherapy. This is owing both to the progressive nature of their PCD lung disease combined with patients’ lack of attendance at outpatient clinic and poor compliance with home treatment: the latter includes daily nebulised rhDNase, rotating prophylactic oral antibiotics and daily chest physiotherapy. In addition, they are reviewed annually at the Royal Brompton Hospital PCD clinic (London, UK). Progression of lung disease is monitored by repeated lung function testing (spirometry, TLC and DLCO), chest radiographs and CT thoraces. Child C;II:1 initially required regular prophylactic in-patient intravenous antibiotic therapy for the first 2 years of life. However, his bronchomalacia has improved and he is currently treated based on clinical symptoms plus radiological findings.
Transmission electron microscopy and video microscopy
Nasal brushings from the five children were analysed at a centre of excellence for PCD (Royal Brompton Hospital London) as previously described.6 Ciliary motility was analysed by video microscopy from transnasal brush biopsies. High-speed (500 frames per second) video sequences of the cilia were captured using a MotionPro X4 camera (Lake Image Systems, Tring, UK) on an inverted Nikon Diaphot microscope (Nikon Instruments Europe, Amsterdam, Netherlands).
Whole-exome sequencing
DNA from one affected individual in each family (A;II:2, B;II:13 and C;II:1) was selected for whole-exome sequencing (GATC, Konstanz, Germany). The exonic DNA was enriched with either the SureSelect 38 or 50 Mb Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA), and sequenced on an Illumina HiSeq (GATC). The 100-bp paired-end reads were aligned and variants/indels identified as previously described.7 Assuming an autosomal recessive model, we prioritised variants that were (i) autosomal, (ii) homozygous, (iii) not present in dbSNP130, (iv) absent or present with a frequency <1% in our 50 Irish control exomes, (v) located within a candidate homozygous region and (vi) absent or present with a frequency <1% in the NHLBI Exome Variant Server database. Copy-number variants and exon deletions were identified from the exome sequencing data using an in-house algorithm.
Genotyping and homozygosity mapping
Genomic DNA from the five affected children was extracted from peripheral lymphocytes and genotyped for 1 million single-nucleotide polymorphisms (SNPs) on the Illumina platform (Illumina, San Diego, CA, USA). The homozygous regions (>1 Mb) shared by the affected children in each family were identified independently using HomozygosityMapper.8
Sanger sequence validation and screening of control panel
The RSPH4A c.166dup and CCNO c.258_262dup duplications and the 3.5-kb deletion in DYX1C1 were validated by Sanger sequence analysis (Supplementary Table S1). The DYX1C1 deletion was also confirmed using SNP data (Supplementary Figure S1). A control panel, comprising 200 control chromosomes from the Irish and Irish Traveller populations, was also screened for each mutation by Sanger sequencing. The identified variants have been submitted to the following databases: http://databases.lovd.nl/shared/genes/RSPH4A, http://databases.lovd.nl/shared/genes/DYX1C1, http://databases.lovd.nl/shared/genes/CCNO.
RESULTS
Ciliary analysis
Analysis of nasal brushings in the affected sib-pair in family A showed that all cilia appeared abnormal and were either static or dyskinetic. On overhead views, the cilia were observed to be circling in motion although the pattern was not full and the cilia appeared stiff with no clearing of debris. Transmission electron microscopy (TEM) revealed a transposition defect with the predominant abnormality (22%) being absence of the central pair, although a significant proportion (25–43%) of ciliary cross-sections had a normal 9+2 pattern (Figure 2; Supplementary Table S2). Displacement of one of the peripheral doublets was observed in some cilia. Outer and inner dynein arms were normal. Nasal epithelial strips from family B were completely nude suggesting ciliary aplasia but it is unclear whether this is a primary or secondary effect. A single rootlet was observed, which argues that the absence of cilia may be due to infection at the time ciliary brushings were taken. As a result, the type of ciliary defect in family B is unknown. Light microscopy showed that the cilia in the proband in family C were static. Electron microscopy revealed that, typically, both inner and outer dynein arms were missing.
Exome sequencing and homozygosity mapping
Assuming a homozygous recessive model, novel homozygous-coding variants were identified in each affected child and a comparison was made across families. We found that there was no novel homozygous variant common to all three families suggesting genetic heterogeneity. Although surprising, this genetic finding is supported by the TEM data, which showed that each family has a different type of ultrastructural defect, and it is therefore plausible that each family may have a different disease mutation. Accordingly, SNP homozygosity mapping and exome variant analysis was undertaken for each family independently. In family A, we identified 25 homozygous segments containing 2768 positional candidate genes that were shared by the two affected siblings (Supplementary Figure S2). Exome analysis identified four novel homozygous-coding variants/indels within the shared homozygous regions (Supplementary Table S3 and S4). One of the four candidate mutations is located within a gene that encodes a known component of the cilium and represents the most likely cause of PCD in family A; RSPH4A (NM_001161664.1:c.166dup; p.Arg56Profs*11). RSPH4A encodes a radial-spoke head protein involved in ciliary movement. There are two previous reports of mutations in RSPH4A associated with PCD type 11 (CILD11; MIM#612649).9, 10 RSPH4A mutations reported to date have been associated with a microtubule transposition phenotype, the same ciliary defect observed in the patients in the current study. The frameshift mutation we identified is novel and is located in the first exon of RSPH4A. Sanger sequence analysis confirmed that both affected siblings are homozygous for the RSPH4A c.166dup duplication (Figure 1c). DNA was not available from unaffected family members to test for segregation. The mutation was not present in 200 control chromosomes from the Irish and Irish Traveller populations. In addition to PCD, the affected children in family A have GSD type III. Exome sequencing showed that these children are homozygous for a 1-bp deletion in AGL (NM_000643.2:c.4197del; p.Ala1400Leufs*15), which has been previously reported in GSD type III.11
The two affected siblings from family B share 19 homozygous regions implicating 860 candidate genes (Figure 2b; Supplementary Figure S3). CNV analysis did not identify any variants of interest (data not shown). Exome analysis and variant prioritisation identified three variants within the candidate loci that are shared by both affected siblings: KCNN3 (NM_002249.5:c.239_241del; p.Gln80del), a calcium-activated potassium channel involved in the regulation of neuronal excitability; CCNO (NM_021147.3:c.258_262dup; p.Gln88Argfs*8), a cyclin O gene involved in the cell cycle and DNA repair and whose expression is strongly induced during multiciliogenesis; and CDKN1C (NM_001122631.1:c.479_490del; p.Ala160_Ala163del), a cyclin-dependent kinase inhibitor involved in differentiation of skeletal muscle and alveoli in the lung and which has been implicated in sporadic cancers (Supplementary Table S4). Of the three genes, CCNO is of greatest interest owing to its location within a region of the genome (5q) that appears to have a major role in multiciliated cell differentiation, a process that gives rise to motile cilia in respiratory airways. Sanger sequencing confirmed segregation of the CCNO mutation with PCD in this family (Figure 2a).
In family C, analysis of homozygous segments and exome sequencing identified 8 regions of homozygosity (349 positional candidate genes) and 11 candidate homozygous variants (Supplementary Figure S4; Supplementary Table S4). However, none of the 11 variants were in genes that were likely to cause a ciliopathy. Analysis of the 349 positional candidate genes identified in the mapping study revealed two genes involved in ciliary function; KIF5C and DYX1C1. Although no single-nucleotide variants or indels were identified in either gene, copy-number variant/exon deletion analysis identified a homozygous deletion of ∼3.5 kb in DYX1C1, which has been previously reported in a family with PCD (Figure 3c).12 The deletion was confirmed by SNP genotyping and PCR (Supplementary Figure S1; Supplementary Table S1). SNP genotyping showed that the deletion was not present in 200 control chromosomes from the Irish and Irish Traveller populations.
DISCUSSION
Owing to founder effects and the limited size of the Irish Traveller population (population size=29 573–40 129),13, 14 each disorder tends to be caused by one common homozygous mutation or a few different mutations within the same disease gene.11, 15 We undertook exome sequencing to identify a putative common PCD disease mutation in the Irish Traveller population. Analysis of three Irish Traveller families with PCD showed that there was no homozygous mutation common to all three families. TEM analysis of nasal ciliary brushings from the three families revealed different types of ciliary defects; transposition defects in family A, nude epithelium in family B and inner and outer dynein arm defects in family C. The presence of different ciliary defects in each of the three Irish Traveller families supports the likelihood of different underlying disease genes. This is surprising given the limited population size and is the first disorder in the Traveller population to be associated with >1 disease gene. Given the reported PCD incidence of 1:15 000–30 000, it is surprising to have identified three unrelated Irish Traveller families with PCD in a total population of ∼30 000–40 000. Even more remarkable is the finding that there are at least three distinct mutations causing PCD in the relatively small Traveller population, suggesting that the mutations are more likely to be of recent origin.
Following this realisation, each family was analysed independently using the combined approach of homozygosity mapping and exome sequencing. In family A, we identified a novel frameshift mutation in the radial-spoke head protein RSPH4A (c.166dup; p.Arg56Pro*11) as the likely cause of PCD in this family. First, TEM of cilia from the patients showed absence of the central microtubular pair, a ciliary defect that is consistent with RSPH4A mutations. Second, the patients have normal situs, as has been the case for all the pathogenic RSPH4A mutations reported to date. Third, the frameshift mutation in RSPH4A introduces a premature stop codon at residue 66, resulting in loss of all annotated domains including the radial-spoke domain (PF04712, residues 209–694; PS50313, residues 370–405; PS50313, residues 507–586). The mutant RSPH4A protein is missing 91% (546/601) of amino acids compared with the wild type, and the truncated protein is predicted to undergo non-sense mediated decay (Supplementary Figure S5). Last, the mutation was not present in 200 Irish control chromosomes, dbSNP, the 1000 Genomes project or the NHLBI ESP database, supporting the likelihood that it is a rare disease-causing mutation. There have been two previous reports of non-sense mutations in exons 1 and 3 of RSPH4A in patients with PCD.9, 10 The clinical symptoms and ultrastructural defects in family A in the current study are very similar to the previously reported patients with RSPH4A mutations; recurrent respiratory infections, chronic wet cough, bronchiectasis, nasal symptoms, normal situs and a transposition defect with absence of the central pair.
Analysis of the affected sib-pair in family B identified novel variants in three positional candidate genes; KCNN3, CCNO and CDKN1C. Of the three genes, CCNO is the strongest candidate for a potential PCD disease gene. We identified a 5-bp duplication in exon 1 of CCNO (c.258_262dup; p.Gln88Argfs*8) that was homozygous in both affected siblings but was not present in the homozygous state in healthy family members. The frameshift mutation results in premature protein truncation at residue 95 (73% of wild-type proteins missing) and the mutant protein is predicted to undergo non-sense mediated decay (Supplementary Figure S6). In mice, CCNO is expressed in the olfactory epithelium and naris anterior epithelium, amongst other tissues. CCNO is involved in the cell cycle and DNA repair. Recently, it has been shown that DNA damage can affect primary ciliogenesis.16 Centriole splitting can occur as a general response to DNA damage and the resulting split centrioles give rise to very few cilia.16, 17 TEM of nasal epithelium from these children showed nude epithelium that was absent of cilia. We hypothesise that the frameshift duplication in CCNO, which is involved in DNA repair, results in the accumulation of DNA damage and centriole splitting. In turn, this may lead to the production of very few cilia and would account for the nude nasal epithelium observed in these children. CCNO is also located within the 5q11 locus, which has a complex but critical role in determining the multiciliated phenotype. CCNO is flanked by IDAS (multicilin), a gene whose expression is required to induce multiciliogenesis, and CDC20B, which encodes a protein that is expressed at the base of each cilium. Indeed, the expression of CCNO itself is strongly induced during multiciliogenesis. Therefore, CCNO makes for a tempting novel PCD gene. Further functional analyses and screening of additional PCD patients is required to determine the extent that CCNO may have in ciliopathies.
The affected singleton in the family is homozygous for a deletion that includes exon 7 of the DYX1C1 gene. Analysis of exome sequence reads shows that the deletion may be as large as 3.9 kb (chr15:g.53516430_53520334del), based on the end coordinates for the nearest upstream read and the start coordinates for nearest downstream read. The deletion was confirmed by logR ratio analysis of the SNP genotype data, which shows a deletion of rs7181226 (chr15:g.53516609-53517109) and rs687623 (chr15:g.53518223-53518723) (Supplementary Figure S1). The nearest flanking SNPs that show normal copy number are rs7167170 (chr15:g.53514629-53515129) and cnv4116p2 (chr15:g.53521450). Therefore, both SNP and exome data indicate that the deleted region is a maximum of 3.9 kb. DYX1C1 encodes a neuronal migration factor, which was first associated with susceptibility to dyslexia. However, both DYX1C1 knockout mice and zebrafish have a ciliary phenotype recapitulating that of PCD in humans. There has been one previous report of PCD patients with recessive loss-of-function mutations in DYX1C1. Indeed, one of the patients reported by Tarkar et al12 has the same DYX1C1 deletion that we identified in one of the Irish Traveller patients in the current study (C;II:1). TEM of nasal epithelium from child C;II:1 showed loss both of inner and outer dynein arms, consistent with the ultrastructural defect observed in previously reported patients with mutations in DYX1C1.
In summary, we have shown that, unexpectedly, PCD is a genetically heterogeneous disorder in the Irish Traveller population. Our study has identified three different PCD genes in this population; two previously reported (RSPH4A and DYX1C1) and one novel candidate gene (CCNO). Analysis of ciliary ultrastructure and patient ethnicity can help to determine which of the 29 PCD genes (28 previously reported and CCNO) may be mutated in each patient and should be prioritised for mutation screening (Table 1).
References
Bush A, Cole P, Hariri M et al: Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J 1998; 12: 982–988.
Bush A, O'Callaghan C : Primary ciliary dyskinesia. Arch Dis Child 2002; 87: 363–365.
Jeganathan D, Chodhari R, Meeks M et al: Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J Med Genet 2004; 41: 233–240.
Palmblad J, Mossberg B, Afzelius BA : Ultrastructural, cellular, and clinical features of the immotile-cilia syndrome. Annu Rev Med 1984; 35: 481–492.
Hornef N, Olbrich H, Horvath J et al: DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med 2006; 174: 120–126.
Antony D, Becker-Heck A, Zariwala MA et al: Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum Mutat 2013; 34: 462–472.
Casey JP, McGettigan P, Lynam-Lennon N et al: Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab 2012; 106: 351–358.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P : HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res 2009; 37: W593–W599.
Zietkiewicz E, Bukowy-Bieryllo Z, Voelkel K et al: Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS One 2012; 7: e33667.
Castleman VH, Romio L, Chodhari R et al: Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet 2009; 84: 197–209.
Crushell E, Treacy EP, Dawe J, Durkie M, Beauchamp NJ : Glycogen storage disease type III in the Irish population. J Inherit Metab Dis 2010; 33 (Suppl 3): S215–S218.
Tarkar A, Loges NT, Slagle CE et al: DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet 2013; 45: 995–1003.
Central Statistics Office, Cork, Ireland. Persons, Males and Females Usually Resident and Present in the State on Census Night, Classified by Age Group and Ethnic or Cultural Background: Ethnic or Cultural Background. Central Statistics Office, 2011, Vol. 5.
All Ireland Traveller Health Study Team, School of Public Health, Physiotherapy and Population Science, University College Dublin, Ireland. All Ireland Traveller Healthy Study. Our Geels, 2011.
Murphy AM, Lambert D, Treacy EP, O'Meara A, Lynch SA : Incidence and prevalence of mucopolysaccharidosis type 1 in the Irish republic. Arch Dis Child 2009; 94: 52–54.
Conroy PC, Saladino C, Dantas TJ, Lalor P, Dockery P, Morrison CG : C-NAP1 and rootletin restrain DNA damage-induced centriole splitting and facilitate ciliogenesis. Cell Cycle 2012; 11: 3769–3778.
Saladino C, Bourke E, Conroy PC, Morrison CG : Centriole separation in DNA damage-induced centrosome amplification. Environ Mol Mutagen 2009; 50: 725–732.
Hjeij R, Lindstrand A, Francis R et al: ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am J Hum Genet 2013; 93: 357–367.
Austin-Tse C, Halbritter J, Zariwala MA et al: Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am J Hum Genet 2013; 93: 672–686.
Panizzi JR, Becker-Heck A, Castleman VH et al: CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat Genet 2012; 44: 714–719.
Onoufriadis A, Paff T, Antony D et al: Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet 2013; 92: 88–98.
Knowles MR, Leigh MW, Ostrowski LE et al: Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am J Hum Genet 2013; 92: 99–106.
Merveille AC, Davis EE, Becker-Heck A et al: CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet 2011; 43: 72–78.
Becker-Heck A, Zohn IE, Okabe N et al: The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet 2011; 43: 79–84.
Loges NT, Olbrich H, Becker-Heck A et al: Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am J Hum Genet 2009; 85: 883–889.
Duquesnoy P, Escudier E, Vincensini L et al: Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am J Hum Genet 2009; 85: 890–896.
Omran H, Kobayashi D, Olbrich H et al: Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008; 456: 611–616.
Mitchison HM, Schmidts M, Loges NT et al: Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat Genet 2012; 44: 381–389.
Schwabe GC, Hoffmann K, Loges NT et al: Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat 2008; 29: 289–298.
Pan Y, McCaskill CD, Thompson KH et al: Paternal isodisomy of chromosome 7 associated with complete situs inversus and immotile cilia. Am J Hum Genet 1998; 62: 1551–1555.
Lucas JS, Adam EC, Goggin PM et al: Static respiratory cilia associated with mutations in Dnahc11/DNAH11: a mouse model of PCD. Hum Mutat 2012; 33: 495–503.
Omran H, Haffner K, Volkel A et al: Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am J Respir Cell Mol Biol 2000; 23: 696–702.
Olbrich H, Haffner K, Kispert A et al: Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet 2002; 30: 143–144.
Failly M, Bartoloni L, Letourneau A et al: Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J Med Genet 2009; 46: 281–286.
Pennarun G, Escudier E, Chapelin C et al: Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet 1999; 65: 1508–1519.
Guichard C, Harricane MC, Lafitte JJ et al: Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am J Hum Genet 2001; 68: 1030–1035.
Loges NT, Olbrich H, Fenske L et al: DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet 2008; 83: 547–558.
Mazor M, Alkrinawi S, Chalifa-Caspi V et al: Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet 2011; 88: 599–607.
Wirschell M, Olbrich H, Werner C et al: The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet 2013; 45: 262–268.
Horani A, Druley TE, Zariwala MA et al: Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet 2012; 91: 685–693.
Olbrich H, Schmidts M, Werner C et al: Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet 2012; 91: 672–684.
Davidson AE, Schwarz N, Zelinger L et al: Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2013; 93: 321–329.
Kott E, Duquesnoy P, Copin B et al: Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet 2012; 91: 958–964.
Zariwala MA, Gee HY, Kurkowiak M et al: ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am J Hum Genet 2013; 93: 336–345.
Duriez B, Duquesnoy P, Escudier E et al: A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc Natl Acad Sci USA 2007; 104: 3336–3341.
Thauvin-Robinet C, Thomas S, Sinico M et al: OFD1 mutations in males: phenotypic spectrum and ciliary basal body docking impairment. Clin Genet 2013; 84: 86–90.
Moore A, Escudier E, Roger G et al: RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet 2006; 43: 326–333.
Dry KL, Manson FD, Lennon A, Bergen AA, Van Dorp DB, Wright AF : Identification of a 5' splice site mutation in the RPGR gene in a family with X-linked retinitis pigmentosa (RP3). Hum Mutat 1999; 13: 141–145.
Zito I, Downes SM, Patel RJ et al: RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet 2003; 40: 609–615.
Kott E, Legendre M, Copin B et al: Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am J Hum Genet 2013; 93: 561–570.
Knowles MR, Ostrowski LE, Loges NT et al: Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am J Hum Genet 2013; 93: 711–720.
Moore DJ, Onoufriadis A, Shoemark A et al: Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am J Hum Genet 2013; 93: 346–356.
Acknowledgements
We sincerely thank the participating families for the use of genetic samples and clinical information. We would also like to thank Ms Melissa Dixon for taking and providing the TEM photographs. This work was co-funded by the Children’s Fund for Health, The Fundraising Office for Temple Street Children’s University Hospital, Dublin, Ireland (PAC10175) and a Medical Research Charities Group grant (MRCG/2011/17) from the National Children’s Research Centre (Our Lady’s Children’s Hospital, Ireland) and the Health Research Board (Ireland). Jillian Casey is supported by a Medical Research Charities Group grant (MRCG/2011/17) through the National Children’s Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Casey, J., McGettigan, P., Healy, F. et al. Unexpected genetic heterogeneity for primary ciliary dyskinesia in the Irish Traveller population. Eur J Hum Genet 23, 210–217 (2015). https://doi.org/10.1038/ejhg.2014.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.79
This article is cited by
-
Genetic and protein interaction studies between the ciliary dyslexia candidate genes DYX1C1 and DCDC2
BMC Molecular and Cell Biology (2023)
-
Cyclin-dependent kinases and rare developmental disorders
Orphanet Journal of Rare Diseases (2020)
-
Motile ciliopathies
Nature Reviews Disease Primers (2020)
-
Atypical cyclins: the extended family portrait
Cellular and Molecular Life Sciences (2020)
-
The development and functions of multiciliated epithelia
Nature Reviews Molecular Cell Biology (2017)
