
Abstract
Split-hand/foot malformation (SHFM) is a congenital limb deformity due to the absence or dysplasia of central rays of the autopod. Six SHFM loci have already been identified. Here we describe a Chinese family with autosomal-dominant SHFM1 that has previously been mapped to 7q21.2-21.3. The two affected family members, mother and son, showed deep median clefts between toes, ectrodactyly and syndactyly; the mother also showed triphalangeal thumbs. Exome sequencing and variant screening of candidate genes in the six loci known to be responsible for SHFM revealed a novel heterozygous mutation, c.558G>T (p.(Gln186His)), in distal-less homeobox 5 (DLX5). As DLX5 encodes a transcription factor capable of transactivating MYC, we also tested whether the mutation could affect DLX5 transcription acitivity. Results from luciferase reporter assay revealed that a mutation in DLX5 compromised its transcriptional activity. This is the first report of a mutation in DLX5 leading to autosomal-dominant SHFM1.
Similar content being viewed by others

Introduction
Split-hand/foot malformation (SHFM), also known as ectrodactyly, is a congenital limb-development defect due to the absence of central rays of the autopod and varying degrees of fusion of the remaining digits. It is characterized by deep median clefts of digits on hands and feet, syndactyly, and hypoplasia of phalanges and metacarpal or metatarsal bones. The phenotype varies in different families and in different limbs of the same patient, ranging from mild syndactyly of three or four fingers to severe ‘lobster-claw’-like hands or feet and even deficiency of all limbs.1 The condition may occur as an isolated limb defect or as a component of a syndrome with other organ defects such as hearing loss, mental retardation and cleft lip and palate.2, 3
SHFM is highly heterogeneous. Linkage or cytogenetic analysis of the nonsyndromic forms in humans has revealed six different loci for SHFM1-6 types: chromosome 7q21.2-q21.3 (SHFM1, MIM 183600),4, 5 Xq26 (SHFM2, MIM 313350),6 10q24 (SHFM3, MIM 600095),7, 8, 9, 10 3q27 (SHFM4, MIM 605289),1 2q31 (SHFM5, MIM 606708)11, 12 and 12q13 (SHFM6, MIM 225300).13, 14 SHFM1, 3, 4 and 5 exhibit autosomal-dominant inheritance; SHFM6 exhibits autosomal-recessive transmission; SHFM2 exhibits X-linked inheritance pattern.6, 15, 16
TP63 (also called P63, MIM603273)17 encodes a homolog of the tumor suppressor p5318 and is associated with both isolated and syndromic SHFM4. Intragenic homozygous mutation of WNT10b (MIM 601906), identified by linkage analysis and positional cloning in 2008, produces the autosomal-recessive SHFM6 phenotype.13 While most SHFM1 cases are caused by various chromosomal aberrations such as deletions, inversions, translocations and duplications in a 1.5-Mb key interval on 7q21 encompassing the DLX5 and DLX6 genes,19, 20, 21 SHFM1 in one family was reported to be due to a homozygous mutation of distal-less homeobox 5 (DLX5) (MIM 600028),22 thus following autosomal-recessive inheritance. The limb malformation is also associated with hearing loss in the family.
DLX5 is homologous with Drosophila distal-less (Dll) and belongs to the DLX family of homeodomain transcription factors. It is expressed mainly in early embryonic development and has an important role in limb, craniofacial and head development and sensory organ morphogenesis.23 In addition, DLX5 acts as an oncogene in lymphomas and lung cancers and can transactivate MYC.24
In this study, we investigated the genetic basis of SHFM in a Chinese family with two members exhibiting isolated SHFM. Using exome sequencing combined with variant screening of candidate genes in the six-mapped loci, we identified a novel heterozygous mutation in exon 3 of DLX5 as the pathogenic culprit for the SHFM1 phenotype in the family. Then we analyzed the function of the mutant gene in transcriptional activation of MYC by luciferase assay. Relative luciferase activity was significantly lower in cells transfected with the mutant DLX5 than the wild-type gene.
Materials and Methods
SHFM1 family
The diagnosis of congenital nonsyndromic SHFM in the family was established by detailed clinical and X-ray examinations. After obtaining informed consent, we obtained blood samples from four family members (I1, I2, II2 and II3), including one affected member (II3). The study was approved by the Ethics Committee of Shandong University School of Medicine.
DNA isolation
Genomic DNA was extracted from peripheral blood leukocytes by a standard salting-out method.25
Whole-exome sequencing
DNA from patient II3 was subjected to whole-exome sequencing (Beijing Genome Institute, Shenzhen, China). Exome capture involved use of the SureSelect Human All Exon Kit (Agilent, Santa Clara, CA, USA), and sequencing involved the HiSeq2000 platform (Illumina, San Diego, CA, USA).
PCR and DNA sequencing
On the basis of the reference sequence for DLX5 (NG_009220.1), primer pairs for exon 3 of DLX5 were designed by use of Primer Premier 5.0 (forward, 5′-TCCGAAGATGCCTCCAGT-3′, reverse, 5′-TTACACGCCATTGGGTCG-3′). PCR conditions were predenaturation at 94 °C for 5 min, 35 cycles of denaturation at 95 °Cfor 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 30 s and final extension at 72 °C for 10 min. PCR products were detected by agarose gel electrophoresis, then underwent direct DNA sequencing.
Plasmids
Two luciferase reporter plasmids driven by a MYC promoter (pGL2-XNM and pGL2-SNM) were gifts from Dr Mark D. Minden (Department of Cellular and Molecular Biology, Ontario Cancer Institute, Toronto, Canada).25 The expression plasmid for wild-type human DLX5 (pcDNA3.1-WT DLX5, WT) was from Drs Joseph R. Testa and Jinfei Xu (Fox Chase Cancer Center, Philadelphia, PA, USA), pGL2-SNM includes the total conventional MYC promoter sequences, containing the two major transcription start sites, whereas pGL2-XNM has only one start site.24
Two segments of mutant cDNA for human DLX5 Q186H (with a nucleotide substitution at position 558) were amplified by PCR with the primer sequences forward (F), 5′-CGGGATCCATGACAGGAGTGTTTGACAG-3′ and reverse (R1), 5′-GATCTTTTGTTATGAAACCAGATTT-3′ (near the 5’) (bold and underline represent the mismatched base); forward (F1), 5′-GGTTTCATAACAAAAGATCCAAG-3′ and reverse (R), 5′-CGGAATTCATAGAGTGTCCCGGAGGCCAG-3′ (near the 3’), with wild-type plasmid DNA as a template. The expression plasmid for Q186H mutant-type human DLX5 (pcDNA3.1-Mut DLX5, Mut) was generated by cloning the mutant cDNA of human DLX5 into EcoRI and BamH1 (Takara, Dalian, China) of pcDNA3.1B (Invitrogen, Shanghai, China). The expression plasmid for another mutant site Q178P(pcDNA3.1-MT DLX5 178P, abbreviated as MT178P) was generated by using the same method.
Bioinformatics analysis
Protein modeling was based on recent data for the DLX5 DNA-binding domain structure in the Protein Data Bank (PDB ID codes 2DJN, http://www.pdb.org).
Cell culture and transfection
HEK293 and HeLa cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin and maintained at 37 °C in a humidified atmosphere of 5% CO2.
Before transfection, 1.6 × 104 HEK293 and 1.8 × 104 HeLa cells were plated into each well of 96-well culture plates and incubated for 24 h. Plasmids (200 ng each) were co-transfected into wells with PCMV-Renilla luciferase as a control (200 ng) by use of Lipofectamine 2000 (200 ng each, Invitrogen). For synchronization, all cells were starved in DMEM without any FBS for 6 h. After starvation, DMEM containing 10% FBS was added to culture plates for an additional 48 h, then cells were split for luciferase detection.
Luciferase reporter assay
HEK293 and HeLa cells at approximately 80 to 90% density were washed with phosphate-buffered saline, then collected in 100 μl 1 × passive lysis buffer and centrifuged for 30 min in the dark at 4 °C. Luciferase activity was detected in the cell lysis solution by use of a dual luciferase reporter assay system (Thermo, USA).
Statistical analysis
Statistical analyses involved two-tailed Student t test. P<0.05 was considered statistically significant.
Results
Clinical features of the family
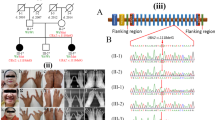
The pedigree of the Chinese family with SHFM is shown in Figure 1. The proband is a 31-year-old woman with typical deep median longitudinal clefts between her toes since she was born (Figure 2a). Her hands appear almost normal, without features of central rays, ectrodactyly, syndactyly or dysplasia of phalanges and metacarpals. However, her thumbs are longer than normal (Figure 2b). X-ray examination revealed absence of the second metatarsals and the second and third toes (Figure 2c); triphalangeal thumbs were also observed (Figure 2d). The second affected member is the 7-year-old son of the proband. Lobster-claw-like feet were present at birth. Other than the split feet, the boy had no other visible abnormalities. None of the remaining three family members, including the parents and the sister of the index person, showed limb development malformations (Figures 2e–h).

Pedigree of the Chinese family with autosomal-dominant split-hand/foot malformation (SHFM). The index patient is indicated by an arrow. I, II, III: first, second, third generation, respectively.

Photographs of hands and feet of family members. (a) The index patient (II 3) showed ‘lobster-claw’-like feet and (b) almost normal hands with slightly longer thumbs than normal. (c) Ectrodactyly and syndactyly in the feet of patient II 3 and (d) triphalangeal thumbs shown on X-ray. (e, f) Normal limbs of father and mother (g, h) of patient II 3.
Identification of a missense mutation in DLX5
Whole-exome sequencing of a DNA sample from patient II3 generated about 6.67 Gb of sequence data, with 96.04% of regions covered at least 4 times and 91.29% of regions covered at least 10 times. Single-nucleotide polymorphisms (SNPs) were filtered through the dbSNP137, 1000 Genomes Project and HapMap8 databases. In all, 1231 SNPs and 115 indels remained. Then we performed variation screening of the 17 candidate genes in the six identified SHFM loci. Finally, we confirmed that a missense mutation in exon 3 of DLX5 (NM_005221.5; c.558G>T, p.(Gln186His)) was present in the affected family members. In addition, the exome sequencing results did not find any coding variants in any other SHFM-associated genes such as TP63, DLX6 and WNT10B.
Direct DNA sequencing revealed the mutation in the index family member but not in all three unaffected family members including healthy parents and 200 ethnically matched healthy controls (Figure 3), which demonstrates that the mutation co-segregated with SHFM in this family and first occurred in the proband.

Sequencing results of exon 3 in DLX5(5′-3′). (a) A heterozygous mutation (G>T) in the index patient (II 3) is indicated by a red arrow. The family members without limb malformation (b) and healthy controls (c) did not carry this mutation.
Alignment of DLX5 amino-acid sequences in different species, including mouse, rat, chimpanzee and opossum, with use of ClustalW2 (http://www.ebi.ac.uk/Tools/ msa/clustalw2/), showed high conservation of glutamine at position 186 (Figure 4a). We analyzed the possible effects of amino-acid substitution of p.(Gln186His) on protein function. The highest possible pathogenicity score (1.0) by polymorphism phenotyping (PolyPhen-2)26 and the lowest tolerance score (0) on sorting intolerant from tolerant (SIFT)27 showed this change as likely damaging protein function. In addition, we found that the mutation is located in the DNA-binding domain of DLX5 and expected to impair DNA binding (Figure 4b). This mutation has already been submitted to dbSNP database.

Bioinformatic analysis of DLX5 mutation (a) Q178 and Q186 are evolutionarily conserved among species. (b) Q186 is located in the DNA-binding domain of DLX5.
DLX5 mutation reduced transcriptional activity
Because human DLX5 is highly expressed in lung and lymph cancers and could bind to two sites in the MYC promoter to activate its transcription,24 we determined the transcriptional activity of the mutant DLX5 (p.(Q186H)) by luciferase reporter assay. In HEK293 cells, compared with transient transfection of pGL2-SNM and pcDNA3.1-WT DLX5, transfection with pcDNA3.1-Mut DLX5 decreased MYC expression by 1.6-fold. Co-transfection of pGL2-XNM with pcDNA3.1-Mut decreased the expression 1.4-fold comparing with pcDNA3.1-WT DLX5 (Figure 5). In HeLa cells, transfection of pGL2-SNM or pGL2-XNM with pcDNA3.1-Mut or pcDNA3.1-WT DLX5 decreased MYC expression by approximately 2- or 3.5-fold, respectively (Figure 5a). Thus, the p.(Q186H) mutation in DLX5 could significantly reduce the transactivation activity of DLX5 (all P<0.05).

Regulation of MYC expression by DLX5 in HEK293 and HeLa cells. (a) Luciferase assay for DLX5 Q186H. SNM and XNM: luciferase reporter plasmids driven by a MYC promoter (pGL2-XNM and pGL2-SNM). WT: expression plasmid of wild-type cDNA of human DLX5 (pcDNA3.1-WT DLX5). Mut: expression plasmid of mutant cDNA of human DLX5 186H (pcDNA3.1-Mut DLX5). Luciferase activity is relative to that of Renilla luciferase. (b) Luciferase assay for Q186H and Q178P in HeLa cells. WT+MT (186H): cotransfection with wide-type DLX5(WT) and 186H mutant plasmid. WT+MT (178P): cotransfection with wide-type DLX5(WT) and 178P mutant plasmid. Data are mean±SD. ***P<0.001.
As DLX5 Q178P has been already reported in a consanguineous autosomal recessive SFMH1 family, to compare the functional effect of the Q178P and Q186H on transactivation activity, we also constructed the expression plasmid for DLX5 Q178P and performed the luciferase assay. However, no difference was found between MT186H and MT178P (Figure 5b). Considering that DLX5 Q186H causes an autosomal dominant SFMH, to test whether this mutation has a dominant negative effect, we conducted the luciferase assay in double transfection with wide-type DLX5(WT) and DLX5 MT186H or MT178P, respectively; no difference was found between WT+MT186H and WT+MT178P, suggesting that these two mutations have similar effects on the gene function in this experiment.
Discussion
SHFM disorders are highly heterogeneous, with variable clinical manifestations, multiple loci and different inheritance modes. Among the four autosomal-dominant forms, SHFM1 was mapped to 7q21.2-21.3 by cytogenetics. Almost all the reported isolated and syndromic cases of SHFM1 have been related to chromosome aberrations, including deletion, duplication, translocation and inversion. However, in one family with syndromic SHFM1, the disease was inherited in an autosomal-recessive mode22 and was caused by a homozygous mutation in exon 2 of DLX5 (c.533A>C), with a glutamine residue at position 178 replaced by a mutant residue proline (p.Q178P). The twin sisters in the family both showed severe limb deformities, with deep median clefts in the hands, ectrodactyly, syndactyly, circumferential nails and asymmetric short limbs as well as hearing loss, low anterior hair line and mild frontal bossing. This was the first identification of an intragenic DLX5 mutation and establishment of a pathogenic gene in SHFM1 syndrome.
The nonsyndromic SHFM1 in the family we described was inherited in an autosomal-dominant manner and was due to a heterozygous mutation in DLX5. The SHFM1 phenotype in this family did not vary greatly. Deep median clefts in the feet and syndactyly were both observed in the two affected family members. In contrast to reported cases of SHFM1 in which the hands of the affected members showed typical lobster-claw deformity, synpolydactyly or monodactyly, the hands in our affected members were nearly unaffected, except that the index person showed only triphalangeal thumbs. Triphalangeal thumbs is probably a rare SHFM phenotype; only one patient with monodactyly combined with triphalangeal thumbs has been reported.2
We performed exome sequencing and identified an intragenic DLX5 missense mutation (c.G558>T), wherein the glutamine residue at position 186 in the wild-type was replaced by histidine. The following lines of evidence indicate that this mutation is the disease-causing culprit. First, DNA sequencing for all four family members revealed that the mutation co-segregated with SHFM in this family. Second, the mutation was not detected in 200 ethnically matched healthy controls, thus ruling it out as a rare SNP. Third, analysis of the effect of this amino-acid substitution on protein function using PolyPhen-2 and SIFT revealed that this mutation was probably damaging. Fourth, the residue at position 186 is highly conserved across species. Finally, because human DLX5 is highly expressed in lung and lymph cancers and can bind to two different sites in the MYC promoter to activate its transcription, we used luciferase reporter assay to determine effect of the mutation c.558G>T on the ability of DLX5 to regulate the expression of MYC. We observed that the mutation significantly reduce the expression of MYC, demonstrating that c.558G>T(p.(Q186H)) affects the ability of DLX5 to regulate transcription of target genes.
The family being studied is the second to be described with an intragenic mutation in DLX5. Although the two families with DLX5 mutations differ in many aspects, including transmission modes, phenotype, expressivity and mutation site, both mutations (Q178P, Q186H) in the DLX5 of these families consisted of the glutamine residue replaced by another residue. In addition, both mutations are in close proximity with each other and localized within the homeodomain of DLX5. Mutations may affect gene function by altering cellular localization, spatial structure of proteins or the recognition site of target genes. Given that facts that Q186H and Q178P mutation in DLX5 cause SHFM with different inheritance pattern, we hypothesized that Q186H might have dominant negative effect, while Q178P is a loss of function mutation. However, our luciferase assay did not give the expected results, these two mutations showed similar functional effects on MYC promoter. We speculated that perhaps there is a second mutation in our studied family. Indeed, Q186H is the first identified heterozygous DLX5 mutation in SHFM, all other mutations identified in man and mouse affect either one copy of two genes (DLX5 and DLX6) or two copies of one gene (DLX5).21, 22 In addition, a recent paper demonstrated that Dlx5 and Dlx6 act cooperatively in the expression of target genes during limb development.28 It is possible that there is another mutation in the regulatory element in the patients of this family.21, 22 Defining the exact pathogenetic mechanism conferred by these mutations requires further investigation.
DLX5 is a member of the DLX gene family, consisting of highly conserved homeobox genes encoding a set of transcription factors that are mainly co-expressed in the median plane of the apical ectodermal ridge (AER) and underlying progress zone of the developing limb bud.29 Complex interaction among diverse molecules involved in BMP or WNT signaling pathways have an important role in maintaining the AER, which is required for limb development in the proximo-distal direction and formation of digits. In mouse models of exsected AER, increased cell death or impaired cell proliferation is involved in inactivating the AER region, which causes SHFM.30, 31 Moreover, many genes, including SHFM disease-causing genes such as TP63 and WNT10b, are expressed in this region.32, 33
DLX5 and DLX6 are very similar in that, both are candidate genes for SHFM1, tandemly arranged on locus 7q21 tail to tail and are expressed in the median AER of embryonic limb buds. Simultaneous knockout of DLX5 and DLX6 in the mouse results in the SHFM phenotype. In contrast, knocking out either one or double heterozygous knockout of DLX5 and DLX6 did not result in limb malformation.4 However, humans are more sensitive than the heterozygous mouse to the dosage of DLX5 and DLX6.4, 34 This situation may explain why the heterozygous point mutation we identified in DLX5 could lead to the typical split-foot malformation in our Chinese family with SHFM.
In summary, this study identified a novel DLX5 mutation in a Chinese family with autosomal-dominant SHFM, which provides additional evidence for the role of DLX5 in limb development.
References
van Bokhoven H, Hamel BC, Bamshad M et al: p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet 2001; 69: 481–492.
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P : Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 2000; 67: 59–66.
Brunner HG, Hamel BC, Van Bokhoven H : The p63 gene in EEC and other syndromes. J Med Genet 2002; 39: 377–381.
Robledo RF, Rajan L, Li X, Lufkin T : The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev 2002; 16: 1089–1101.
Merlo GR, Paleari L, Mantero S et al: Mouse model of split hand/foot malformation type I. Genesis 2002; 33: 97–101.
Faiyaz-Ul-Haque M, Zaidi SH, King LM et al: Fine mapping of the X-linked split-hand/split-foot malformation (SHFM2) locus to a 5.1-Mb region on Xq26.3 and analysis of candidate genes. Clin Genet 2005; 67: 93–97.
de Mollerat XJ, Gurrieri F, Morgan CT et al: A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 2003; 12: 1959–1971.
Ozen RS, Baysal BE, Devlin B et al: Fine mapping of the split-hand/split-foot locus (SHFM3) at 10q24: evidence for anticipation and segregation distortion. Am J Hum Genet 1999; 64: 1646–1654.
Roscioli T, Taylor PJ, Bohlken A et al: The 10q24-linked split hand/split foot syndrome (SHFM3): narrowing of the critical region and confirmation of the clinical phenotype. Am J Med Genet A 2004; 124A: 136–141.
Kano H, Kurosawa K, Horii E et al: Genomic rearrangement at 10q24 in non-syndromic split-hand/split-foot malformation. Hum Genet 2005; 118: 477–483.
Del Campo M, Jones MC, Veraksa AN et al: Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am J Hum Genet 1999; 65: 104–110.
Goodman FR, Majewski F, Collins AL, Scambler PJ : A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am J Hum Genet 2002; 70: 547–555.
Ugur SA, Tolun A : Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot Malformation. Hum Mol Genet 2008; 17: 2644–2653.
Khan S, Basit S, Zimri FK et al: A novel homozygous missense mutation in WNT10B in familial split-hand/foot malformation. Clin Genet 2012; 82: 48–55.
Ahmad M, Abbas H, Haque S, Flatz G : X-chromosomally inherited split-hand/split-foot anomaly in a Pakistani kindred. Hum Genet 1987; 75: 169–173.
Faiyaz ul Haque M, Uhlhaas S, Knapp M et al: Mapping of the gene for X-chromosomal split-hand/split-foot anomaly to Xq26-q26.1. Hum Genet 1993; 91: 17–19.
Celli J, Duijf P, Hamel BC et al: Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999; 99: 143–153.
Yang A, Kaghad M, Wang Y et al: p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Scherer SW, Poorkaj P, Allen T et al: Fine mapping of the autosomal dominant split hand/split foot locus on chromosome 7, band q21.3-q22.1. Am J Hum Genet 1994; 55: 12–20.
Crackower MA, Scherer SW, Rommens JM et al: Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3-q22.1 and analysis of a candidate gene for its expression during limb development. Hum Mol Genet 1996; 5: 571–579.
Velinov M, Ahmad A, Brown-Kipphut B et al: A 0.7 Mb de novo duplication at 7q21.3 including the genes DLX5 and DLX6 in a patient with split-hand/split-foot malformation. Am J Med Genet A 2012; 158A: 3201–3206.
Shamseldin HE, Faden MA, Alashram W, Alkuraya FS : Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. J Med Genet 2012; 49: 16–20.
Merlo GR, Zerega B, Paleari L, Trombino S, Mantero S, Levi G : Multiple functions of Dlx genes. Int J Dev Biol 2000; 44: 619–626.
Xu J, Testa JR : DLX5 (distal-less homeobox 5) promotes tumor cell proliferation by transcriptionally regulating MYC. J Biol Chem 2009; 284: 20593–20601.
Han Y, San-Marina S, Liu J, Minden MD : Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene 2004; 23: 6933–6941.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Vieux-Rochas M, Bouhali K, Mantero S et al: BMP-mediated functional cooperation between Dlx5;Dlx6 and Msx1;Msx2 during mammalian limb development. PLoS One 2013; 8: e51700.
Ferrari D, Harrington A, Dealy CN : Kosher RA: Dlx-5 in limb initiation in the chick embryo. Dev Dyn 1999; 216: 10–15.
Duboule D : Making progress with limb models. Nature 2002; 418: 492–493.
Saunders Jr JW : Is the progress zone model a victim of progress? Cell 2002; 110: 541–543.
Shimomura Y, Wajid M, Shapiro L, Christiano AM : P-cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development 2008; 135: 743–753.
Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S : Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns 2009; 9: 215–223.
Elliott AM, Evans JA : Genotype-phenotype correlations in mapped split hand foot malformation (SHFM) patients. Am J Med Genet A 2006; 140: 1419–1427.
Acknowledgements
We thank the family members for participating in our study, and Professor Joseph R Testa (Fox Chase Cancer Center, Philadelphia, PA) and Dr Mark D Minden (Department of Cellular and Molecular Biology, Ontario Cancer Institute, Toronto, Canada) for plasmids. This work was supported by grants from the Natural Science Foundation of China (81072452, 81273281) and Ph.D. Programs Foundation of Ministry of Education of China (20100131110035).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Wang, X., Xin, Q., Li, L. et al. Exome sequencing reveals a heterozygous DLX5 mutation in a Chinese family with autosomal-dominant split-hand/foot malformation. Eur J Hum Genet 22, 1105–1110 (2014). https://doi.org/10.1038/ejhg.2014.7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.7
Keywords
This article is cited by
-
Limb development: a paradigm of gene regulation
Nature Reviews Genetics (2017)
-
Phenotypic subregions within the split-hand/foot malformation 1 locus
Human Genetics (2016)
-
Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: array CGH screening of 134 unrelated families
Orphanet Journal of Rare Diseases (2014)
