
Abstract
Phylogenetically informative Y chromosomal single-nucleotide polymorphisms (Y-SNPs) integrated in DNA chips have not been sufficiently explored in most genome-wide association studies (GWAS). Herein, we introduce a pipeline to retrieve Y-SNP data. We introduce the software YTool (http://mitotool.org/ytool/) to handle conversion, filtering, and annotation of the data. Genome-wide SNP data from populations in Myanmar are used to construct a haplogroup tree for 117 Y chromosomes based on 369 high-confidence Y-SNPs. Parallel genotyping and published resequencing data of Y chromosomes confirm the validity of our pipeline. We apply this strategy to the CEU HapMap data set and construct a haplogroup tree with 107 Y-SNPs from 39 individuals. The retrieved Y-SNPs can discern the parental genetic structure of populations. Given the massive quantity of data from GWAS, this method facilitates future investigations of Y chromosome diversity.
Similar content being viewed by others

Introduction
The non-recombining portion of the Y chromosome is the most genealogically informative haploid marker in the human nuclear genome.1 In 2002, the Y Chromosome Consortium constructed a Y chromosomal haplogroup tree based on 245 biallelic single-nucleotide polymorphisms (Y-SNPs) including indels that were generally treated as binary SNPs. Analyses of these data defined 153 human Y chromosomal haplogroups.2 Subsequently, the Y chromosome tree was modified1, 3 and updated.4 The updated tree provides an important foundation for studies of evolutionary anthropology,5 genealogical reconstruction,6 molecular forensics,7 and medical genetics.8
Current commercial DNA chips (microarrays) for massive genome-wide association studies (GWAS)9 are designed to contain many Y-SNPs. In 2007, Underhill and Kivisild retrieved 295 Y-SNPs from customized Perlegen arrays in a previous study10 and used them to reconstruct the Y chromosome tree for 33 males.3 Their work suggested that Y-SNP data from DNA chips—usually the byproduct of GWAS—provide opportunities to explore Y chromosome diversity within and among populations. Although Y-SNP data from DNA chips helped improve assessments of population stratification,11 many GWAS do not consider or evaluate Y-SNPs. It is difficult to tease out Y-SNPs from bulk GWAS data and then to assign the Y chromosome to a haplogroup.
Herein, we develop a de novo pipeline for retrieving Y-SNPs in DNA chips. We test our approach using data of DNA chips from populations in Myanmar (also known as Burma). Only sporadic sampling exists for Myanmar12 and, thus, a systematic investigation of the Y chromosome diversity is wanting. In addition to providing a practical protocol of analyzing Y-SNPs from GWAS data, our work adds an essential piece to the genetic puzzle for Southeast Asians.
Materials and methods
Sample collection
We collected blood samples from 106 unrelated male individuals living in Myanmar (Table 1). Four different ethnic groups were surveyed: Bamar (also Burman, n=59), Chin (n=19), Naga (n=15), and Rakhine (n=13). Blood samples of eight unrelated individuals of the Jingpo ethnic group from Yunnan, China, were collected to represent the Kachin people of northern Myanmar because the two names appear as synonyms for the same ethnic group.13 Blood samples of three Nigerian males were used as the outgroup reference. All 117 subjects were interviewed to obtain informed consent before sample collection. The Institutional Review Board of the Kunming Institute of Zoology approved the protocols and study.
DNA extraction and genome-wide SNP genotyping
Genomic DNA was extracted and purified by using QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Genome-wide SNPs for each sample were genotyped with HumanOmniZhongHua-8 BeadChip (http://support.illumina.com/array/array_kits/humanomnizhonghua-8_beadchip_kit.ilmn; Illumina, San Diego, CA, USA) according to the manufacturer’s protocols. The 900015 markers on this chip captured 81% variation (r2>0.8) with minor allele frequency >5% in East Asians (CHB+JPT). Given the close relationship between East and Southeast Asians,12, 14 we used the HumanOmniZhongHua-8 BeadChip to investigate Myanmarese populations. Compared with the contemporary Illumina HumanOmni1-Quad BeadChip (1140419 markers in total, including 2322 Y-SNPs), the HumanOmniZhongHua-8 BeadChip provided a considerable quantity of markers (900015, including 2041 Y-SNPs) at a much lower cost.
Data sets
Genome-wide SNPs in the HumanOmniZhongHua-8 BeadChip were scanned by iScan (Illumina). The chip data with PLINK format (.MAP and.PED files) were exported through GenomeStudio (Illumina). The alleles were mapped to the forward strand. Excluding pseudoautosomal region loci coded by chromosome 25, we extracted data for 2041 Y-SNPs in the male-specific region (coded by chromosome 24) of 117 males using PLINK 1.07 (http://pngu.mgh.harvard.edu/∼purcell/plink/).15
Data filtering
Owing to the unusual nature of Y chromosomal characters, genotyping of human Y-SNPs using DNA chips has been problematic.10 The high level of similarity between certain regions of Y and X chromosomes/autosomes16, 17 can blur signals of genotyping Y-SNPs, as can the very high proportion of ampliconic repeats that are often highly similar to each other (usually >99.9% identity).16, 17 Consequently, we employed specific quality controls and excluded the following data in 117 male samples: (1) Y-SNPs with heterozygous alleles; (2) Y-SNPs genotyped with a missing rate more than 5%; and (3) known Y-SNPs with identical genotypes (ie, invariants).
Phylogenetic analyses
We constructed median-joining network18 with the filtered Y-SNPs by using NETWORK 4.611 (http://www.fluxus-engineering.com/sharenet.htm) to infer the genealogy for each of Y chromosomes. Each Y-SNP was conveniently mapped on branches of the genealogy during network construction. The Y chromosome haplogroup tree was constructed from the genealogy. To reconcile our results with the widely accepted Y chromosomal phylogeny and haplogroup definitions,4 we consulted Y-DNA Haplogroup Tree 2013 of ISOGG (http://www.isogg.org/tree/),19 which was based on Karafet et al4 but constantly updated. We followed the previously used nomenclature (ie, lineage-marker) for each haplogroup proposed by Karafet et al4 To automatically address the data conversion, filtering, and annotation, we also developed C++ stand-alone software with GUI: YTool (http://mitotool.org/ytool/; Supplementary Methods). YTool was also designed to export Y-SNP data in the FASTA format for use in alternative software.
Validation
Two parallel strategies were adopted to validate the DNA chip data. First, in terms of the known Y chromosome tree,4 28 diagnostic Y-SNPs defining the common Y chromosomal haplogroups (M168-CT, M145-DE, M174-D, M55-D2, M69-H, M410-J2a, M241-J2b2, M11-L, P188-NO, P191-O, P203.1-O1a, M50-O1a2, P31-O2, M95-O2a, M88-O2a1, M122-O3, P199-O3a, P201-O3a3, M134-O3a2c1, M117-O3a2c1a, M162-O3a2c1a1, M120-Q1a1, M207-R, P231-R1, M198-R1a1a, M87-R1a1a1c, M269-R1b1b, and M124-R2a) in Southeast Asia20, 21 and Northeast India22 were selected. These Y-SNPs were amplified in three panels of multiplex PCR reactions (Supplementary Methods) and then were genotyped with GenomeLab SNPstream (Beckman Coulter, Fullerton, CA, USA). Based on our previous studies,20, 22 primers for multiplex PCR and single base extension reactions were designed by Autoprimer software (Beckman Coulter). Further sequencing with an ABI 3730 DNA Analyzer (Applied Biosystems by Life Technologies, Foster City, CA, USA) was used to confirm specific Y-SNPs. The PCR amplification and sequencing primers were previously reported.4 In terms of the diagnostic Y-SNPs, we classified each of the 117 males into their known haplogroup, and then compared the results with those based on DNA chips. Second, we consulted the 6662 Y-SNPs reported in a recent study of Y chromosome using next-generation sequencing (ie, NGS data).23 We compared our results with the NGS data, especially by checking the annotation of phylogenetic status for each of the Y-SNPs.
Application
We downloaded Y-SNPs of CEU (ie, genotypes_chrY_CEU_r28_nr.b36_fwd.txt.gz) from HapMap (ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/2010-08_phaseII+III/forward/). Because the samples were genotyped using different platforms, we only considered 44 individual males investigated by all chips. The data were transformed into PLINK format. Five individuals with high proportions of missing genotypes (>61%) were excluded. Finally, we tested the data of 943 Y-SNPs in 39 CEU males with our method implemented in YTool. All sample data as well as results for analyses in this work were made available at http://mitotool.org/ytool/ytool.zip.
Results
Data filtering
Call rates for each sample were above 98% for the HumanOmniZhongHua-8 BeadChips. For the 2041 Y-SNPs (Supplementary Data 1 and 2), the proportions of missing genotypes varied from 10.3– to 20.4% (11.3%, on average). First, 241 Y-SNPs (∼11.8%, 241/2041) genotyped with heterozygous alleles in the 117 male samples were disregarded. Second, 79 Y-SNPs (∼3.9%, 79/2041) with missing genotypes in more than 5% of the male samples were excluded. Third, 1346 Y-SNPs (∼65.9%, 1346/2041) identified as invariant were not considered. As a result of this filtering, 375 variants were remained (Table 2). Y-SNPs filtered in each step were deposited in downloadable files (http://mitotool.org/ytool/ytool.zip).
Y chromosomal haplogroup tree
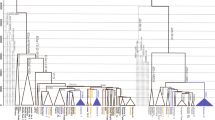
Of the 375 Y-SNPs for 117 males, six Y-SNPs occurred multiple times on different branches of the median-joining network. These likely represented multiple mutational events. Because recurrent mutations are rare2, 4, 23 and should be treated with caution,24, 25 we took a conservative approach and excluded these six Y-SNPs from subsequent analyses. Thus, we reconstructed the Y chromosomal haplogroup tree based on 369 phylogenetically informative Y-SNPs (Figure 1). According to this tree, all samples were assigned into 22 (sub-)haplogroups (paragroups) within macrohaplogroup CT (Supplementary Table 1).

Y chromosomal haplogroup tree of 117 male samples. Reference sequence numbers (rs#) of the Y-SNPs defining the tree structure are shown along the branches. Population codes are given in Table 1 followed by sample sizes for each of the haplogroups. Y-SNPs without rs# are noted in italics: 610147 (200610-147), 610155 (200610-155), 610158 (200610-158), 610191 (200610-191), 610195 (200610-195), 610214 (200610-214), 610226 (200610-226), 610245 (200610-245), 610281 (200610-281), 610363 (200610-363), 610366 (200610-366), 8Y2897 (2010-08-Y-2897), and 8Y3175 (2010-08-Y-3175). Y-SNPs are annotated with their corresponding markers deposited in ISOGG (http://www.isogg.org/tree/ISOGG_YDNA_SNP_Index.html). Y-SNPs identified in NGS data are in red, whereas those out of nine YSURs are in blue (Table 2).23 Those distributing in YSURs but not found in NGS data are in black. Y-SNPs genotyped by SNPstream or direct sequencing are underlined. Problematic Y-SNPs are given in parentheses.
Validation
The phylogeny of Myanmarese Y chromosomal lineages was largely supported by genotyping and direct sequencing the candidate diagnostic Y-SNPs (Figure 1; Supplementary Table 2). There was one conflict because of variant rs34893929 (ie, Page23) detected in the branch of haplogroup O3a2c-8Y2897 (Figure 1). Page23 is equivalent to M117 defining haplogroup O3a2c1a-M117, which was a downstream diagnostic variant of M134 defining O3a2c1-M134.19 However, both M117 and M134 were absent in our samples (Supplementary Table 2). Thus, genotyping of Page23 was a false positive signal. Similarly, variant rs34441307 (ie, L137), which defined haplogroup I2a2a1a1-L13719 and occurred on branch O3*-M122, was likely a phantom.
To validate the remaining Y-SNPs in the chip, we referenced the 6662 Y-SNPs identified in a recent resequencing study of nine Y-specific unique regions (YSURs) spanning 8.97 Mb in which pseudoautosomal, heterochromatic, X-transposed, and ampliconic segments were excluded.23 Comparison of two data sets was shown in Table 2. About 50.4% (842/1672) of the Y-SNPs removed in our data filtering occurred out of the YSURs. In contrast, only ∼20.3% (75/369) of the Y-SNPs passing through our filtering distributed out of the YSURs. The difference was statistically significant (χ2test; p<0.0001; Table 3). Further, no conflicts were found upon checking the phylogenetic status for each of 244 variants annotated in NGS data (Figure 1). For the branches investigated in both studies (eg, CF-F-IJK-P-R-R1), almost all variants distributed within the YSURs were confirmed by NGS data, except for rs2049642 in the branch directing to haplogroup IJK (Figure 1). Because the NGS data were from 36 males only representing haplogroups A, D, E, G, I, N, Q, and R, Y-SNPs for other haplogroups (eg, C, H, J, L, and O) were not evaluated.23
Application: CEU in HapMap
The data filtering for 943 Y-SNPs in 39 males was shown in Table 2. The Y chromosomal haplogroup tree based on 107 qualified Y-SNPs was constructed (Figure 2). The phylogeny was generally consistent with the annotation of ISOGG and NGS data. All 39 males were assigned into two major European Y chromosome haplogroups I and R1, suggesting a European ancestry. Three samples (NA06994, NA07357, and NA12891) that were both genotyped by chips and sequenced by NGS were concordant (Figure 2).

Y chromosomal haplogroup tree of 39 CEU males in HapMap. The phylogenetic status of seven Y-SNPs cannot be determined, and these Y-SNPs are shown in box with dashes. For the information of the labels, see Figure 1 and its legend.
Discussion
We provide a practical strategy to retrieve informative Y-SNPs from GWAS data using YTool software. The software also exports retrieved Y-SNPs into FASTA format for further analysis by many phylogenetic tools. Both traditional genotyping and published NGS data generally validate our method. Our pipeline can be used to discern data quality, mutational stabilities, and phylogenetic status of multiple populations involved massive GWAS. The Y chromosomal haplogroup tree can be constructed (eg, Figure 1) and the haplogroup profiles of populations can be depicted (eg, Supplementary Table 1). Further analyses, such as principal component analysis26 and analysis of molecular variance,27 are able to clarify the parental genetic structures.
The Y chromosomal haplogroup tree retrieved from GWAS data is valuable for exploring population genetic diversity. Most studies rely on genotyping 20–30 candidate diagnostic Y-SNPs in Southeast Asians (eg, He et al20). In contrast, resolution of the Y chromosomal haplogroup tree for Myanmar is improved by analyzing hundreds of high-confidence variants (Figure 1). In terms of this tree, the future employment of Y-STRs28 promises to reveal more details about parental demographic history in Myanmar.
One should consider the quality of Y-SNP data integrated in DNA chips. Of the 2041 and 943 Y-SNPs retrieved from the HumanOmniZhongHua-8 BeadChip and multiple chips used in CEU, 81.9% (1672/2041) and 88.7% (836/943) Y-SNPs were removed in data filtering, respectively (Table 2). Although we could not exclude inherent genotyping or calling errors in DNA chips, the high proportion of filtered data likely reflect the design of the probes. Around 50.4% (832/1672) and 82.1% (686/836) of filtered Y-SNPs for two respective data sets occur outside of the nine YSURs (Table 3). These filtered Y-SNPs may distribute in the heterochromatic, X-transposed, and ampliconic segments of the Y chromosome. As a result, the specificity of probes in the DNA chips needs to be improved. This can be achieved, at least partially, by replacing the problematic markers with those identified in Y chromosome resequencing (eg, see Wei et al,23 Cruciani et al,29 Xue et al30) and then redesigning the probes. We also suggest that DNA chip manufacturers use our method to test their new products before release.
References
Jobling MA, Tyler-Smith C : The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 2003; 4: 598–612.
Y Chromosome Consortium: A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res 2002; 12: 339–348.
Underhill PA, Kivisild T : Use of Y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet 2007; 41: 539–564.
Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF : New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 2008; 18: 830–838.
Chiaroni J, Underhill PA, Cavalli-Sforza LL : Y chromosome diversity, human expansion, drift, and cultural evolution. Proc Natl Acad Sci USA 2009; 106: 20174–20179.
King TE, Jobling MA : What's in a name? Y chromosomes, surnames and the genetic genealogy revolution. Trends Genet 2009; 25: 351–360.
Kayser M, de Knijff P : Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 2011; 12: 179–192.
Charchar FJ, Bloomer LD, Barnes TA et al: Inheritance of coronary artery disease in men: an analysis of the role of the Y chromosome. Lancet 2012; 379: 915–922.
Fan JB, Chee MS, Gunderson KL : Highly parallel genomic assays. Nat Rev Genet 2006; 7: 632–644.
Hinds DA, Stuve LL, Nilsen GB et al: Whole-genome patterns of common DNA variation in three human populations. Science 2005; 307: 1072–1079.
Makowsky R, Yan Q, Wiener HW et al: The utility of mitochondrial and y chromosome phylogenetic data to improve correction for population stratification. Front Genet 2012; 3: 301.
Chaubey G, Metspalu M, Choi Y et al: Population genetic structure in Indian Austroasiatic speakers: the role of landscape barriers and sex-specific admixture. Mol Biol Evol 2011; 28: 1013–1024.
Wang Z : The Jingpo: Kachin of the Yunnan Plateau. Tempe: Arizona State University, 1997.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Hughes JF, Rozen S : Genomics and genetics of human and primate Y chromosomes. Annu Rev Genomics Hum Genet 2012; 13: 83–108.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ et al: The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003; 423: 825–837.
Bandelt HJ, Forster P, Röhl A : Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48.
International Society of Genetic Genealogy. 2013, Y-DNA Haplogroup Tree 2013. Version: 8.20, Date: 26 February 2013 http://www.isogg.org/tree/ Accessed on: 28 Februray 2013..
He JD, Peng MS, Quang HH et al: Patrilineal perspective on the Austronesian diffusion in Mainland Southeast Asia. PLoS One 2012; 7: e36437.
Karafet TM, Hallmark B, Cox MP et al: Major east-west division underlies Y chromosome stratification across Indonesia. Mol Biol Evol 2010; 27: 1833–1844.
Debnath M, Palanichamy MG, Mitra B, Jin JQ, Chaudhuri TK, Zhang YP : Y-chromosome haplogroup diversity in the sub-Himalayan Terai and Duars populations of East India. J Hum Genet 2011; 56: 765–771.
Wei W, Ayub Q, Chen Y et al: A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res 2013; 23: 388–395.
Adams SM, King TE, Bosch E, Jobling MA : The case of the unreliable SNP: recurrent back-mutation of Y-chromosomal marker P25 through gene conversion. Forensic Sci Int 2006; 159: 14–20.
Cruciani F, Trombetta B, Novelletto A, Scozzari R : Recurrent mutation in SNPs within Y chromosome E3b (E-M215) haplogroup: a rebuttal. Am J Hum Biol 2008; 20: 614–616.
Richards M, Macaulay V, Torroni A, Bandelt HJ : In search of geographical patterns in European mitochondrial DNA. Am J Hum Genet 2002; 71: 1168–1174.
Excoffier L, Smouse PE, Quattro JM : Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 1992; 131: 479–491.
Ballantyne KN, Goedbloed M, Fang R et al: Mutability of Y-chromosomal microsatellites: rates, characteristics, molecular bases, and forensic implications. Am J Hum Genet 2010; 87: 341–353.
Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R : A revised root for the human Y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am J Hum Genet 2011; 88: 814–818.
Xue Y, Wang Q, Long Q et al: Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 2009; 19: 1453–1457.
Acknowledgements
We are grateful to all volunteers and samplers. We thank Zhong-Yin Zhou, Wen-Zhi Wang, and Ni-Ni Shi for technical assistance. This study was supported by grants from National Natural Science Foundation of China and Bureau of Science and Technology of Yunnan Province. The Youth Innovation Promotion Association, Chinese Academy of Sciences provided support to M-SP. This work was also supported, in part, by a Visiting Professorship for Senior International Scientists from the Chinese Academy of Sciences, and manuscript preparation by a Natural Sciences and Engineering Research Council of Canada Discovery Grant A3148 to RWM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Peng, MS., He, JD., Fan, L. et al. Retrieving Y chromosomal haplogroup trees using GWAS data. Eur J Hum Genet 22, 1046–1050 (2014). https://doi.org/10.1038/ejhg.2013.272
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.272
Keywords
This article is cited by
-
Reconstruction of Y-chromosome phylogeny reveals two neolithic expansions of Tibeto-Burman populations
Molecular Genetics and Genomics (2018)
-
No association between Y chromosomal haplogroups and severe acne in the Han Chinese population
Journal of Human Genetics (2014)
