
Abstract
Sardinia has been used for genetic studies because of its historical isolation, genetic homogeneity and increased prevalence of certain rare diseases. Controversy remains concerning the genetic substructure and the extent of genetic homogeneity, which has implications for the design of genome-wide association studies (GWAS). We revisited this issue by examining the genetic make-up of a sample from North-East Sardinia using a dense set of autosomal, Y chromosome and mitochondrial markers to assess the potential of the sample for GWAS and fine mapping studies. We genotyped individuals for 500K single-nucleotide polymorphisms, Y chromosome markers and sequenced the mitochondrial hypervariable (HVI–HVII) regions. We identified major haplogroups and compared these with other populations. We estimated linkage disequilibrium (LD) and haplotype diversity across autosomal markers, and compared these with other populations. Our results show that within Sardinia there is no major population substructure and thus it can be considered a genetically homogenous population. We did not find substantial differences in the extent of LD in Sardinians compared with other populations. However, we showed that at least 9% of genomic regions in Sardinians differed in LD structure, which is helpful for identifying functional variants using fine mapping. We concluded that Sardinia is a powerful setting for genetic studies including GWAS and other mapping approaches.
Similar content being viewed by others

Introduction
Genome-wide association studies (GWAS) have become the standard approach to identify genes involved in multifactorial human traits, with hundreds of genetic variants that have been identified for over 200 human traits.1 Despite this success, only a few variants have actually been proven to be functional.2 The difficulty in identifying the actual causal variant using GWAS is partly due to the presence of linkage disequilibrium (LD), which prohibits high resolution fine mapping, and partly to the underlying genetic architecture of complex traits.2 Therefore, other approaches are needed to identify the causal variants involved in human traits.
Genetically isolated populations are attractive for genetic studies because of their decreased genetic heterogeneity and extended LD.3 In addition, because of founder and drift effects,4, 5 variants that are rare in outbred populations may have an increased frequency and therefore a larger associated genetic risk.4 This is advantageous for GWAS, as the increase in risk makes it easier to identify a variant using a relatively small sample.4, 6 In addition, the increased homozygosity facilitates the identification of recessive variants.7, 8
Sardinia is considered an old genetic isolate, based on historical and archeological records,9 genetic analyses10, 11 and the increased prevalence of certain disorders.12, 13 In this population, several GWAS have already been carried-out.14 Still, controversy remains as to whether Sardinia is a homogeneous genetic isolate. Reports based on Y microsatellite markers and a few autosomal loci have shown that Sardinians from different regions of the island cannot be distinguished by their genetic make-up,15 which implies that pooling individuals from different regions in Sardinia will not lead to substantial population stratification. However, in a recent study, it was shown that even within a single province in Sardinia, there are distinct sub-populations.16
Differences between these findings are difficult to assess because both types of genetic data provide different information about the genetic structure of a population. Markers from the Y chromosome and mitochondrial DNA (mtDNA) are preferred to assess population movements and evaluate past founder events. However, they provide little information regarding LD patterns and minor allele frequencies (MAFs), which are the parameters that are important for GWAS. In addition, most studies analyzing the genetic variation in Sardinians have been performed with small data sets,11, 17 which tend to inflate parameters such as MAF and LD.18 Here, we characterize the genetic make-up of a population sample from North-East (NE) Sardinia, to aid in the design of powerful GWAS. In addition, we assess the population substructure in this region of Sardinia. In addition, we assessed population substructure in this region of Sardinia as it includes the Northern part of the archaic region of Barbagia, considered from historical records the most isolated part of the island.
Subjects and methods
Subjects and geographical distribution of the sample
A sample of 260 males and 46 females from NE Sardinia was selected. All participants were healthy blood donors recruited at the Nuoro Hospital, a major referral hospital in the region. We only included unrelated subjects with Sardinian ancestry for at least two generations, based on self-reported ancestry and municipal records. The participants gave informed consent. The protocol was approved by the local ethical committee.
The geographical distribution of the places of birth of our sample is shown in Figure 1. The villages are distributed throughout six regions: Nuorese, Sassari, Oristano, Baronia, Ogliastra and Marghine-Planargia. Barbagia, the most isolated part of Sardinia, is located within the province of Nuoro. We investigated sub-population differentiation by grouping the regions into the archaic region of Barbagia (Nuorese) and the rest.

Geographical distribution of the places of birth of the Sardinian sample. Nuorese: purple; Sassari: green; Baronia: pink; Ogliastra: deep blue; Marghine-planargia: light blue; Oristano: red.
Genotyping
We selected 260 males for the identification of founder lineages. We determined the variation of the non-recombinant part of the Y chromosome (NRY) using a total of 36 NRY single-nucleotide polymorphisms (SNPs) and 15 microsatellite markers. For the maternal haplotypes, we sequenced the hypervariable segments HVS-I (nucleotides 16001–16568) and HVS-II (nucleotides 001–574) of the mtDNA. For the autosomal data, a sample of 118 individuals, which included 72 males from the Y chromosome sample and 46 females were genotyped using the Affymetrix Genome Wide SNP v5.0 arrays (Affymetrix, High Wycomb, UK). Details on genotyping protocols and quality control are provided in Supplementary Methods. The genotype files are available on request.
Data analysis
Estimation of paternal and maternal lineages and comparison with other populations
We estimated the frequency of major haplogroups from both Y chromosome and mitochondrial systems from 260 males and compared the haplogroup frequencies with estimates from other Mediterranean populations using published reports.19 Standard measures of diversity including haplotype diversity, mean heterozygosity and mean number of alleles were calculated for both Y chromosome and mitochondrial systems using algorithms implemented in ARLEQUIN.20 To assess the degree of differentiation between the Sardinian population and other populations, we estimated pairwise population genetic distance, calculated as the Fst statistic. To estimate population differentiation using the haplotypes from the STR markers, we used Rst. The significance of pairwise Fst and Rst estimates was assessed based on 10 000 permutations. The above analyses of molecular variance and population differentiation were carried-out using the ARLEQUIN software.20
Assessment of stratification, relatedness and homozygosity in the Sardinians
To evaluate the extent of structure in the Sardinian sample, we used the genotypes from the autosomal markers. First, we selected markers in linkage equilibrium (LE) with which to perform principal component analysis (PCA). To prune SNPs that were LE, we used PLINKv2.07 to remove SNPs in high LD (R2>0.8) in windows of 50 SNPs and shifting the windows every 5 SNPs.21 Next, we used the SMARTPCA software from the EIGENSOFT package v.02 for PCA with parameter defaults.22
The set of markers in LE was also used to investigate the degree of relatedness among individuals and the extent of homozygosity. To investigate relatedness, we estimated the average kinship coefficient (π) in all Sardinian pairs, calculated as: π=p[IBD=2]+0.5 × p[IBD=1]. Pairs with π>0.05 are related within six meioses. Further, we evaluated the degree of homozygosity by calculating both the average homozygosity (F) and runs of homozygosity (ROH).The latter were defined as stretches of marker homozygosity of at least 1 Mb, allowing for one heterozygous SNP and one missing SNP, with a minimum of 20 SNPs per window and a maximum gap of 50 kb between windows. We retrieved the mean number of ROH segments per individual, the mean length of the homozygous segment and the total amount of DNA containing the homozygous regions. The above analyses were performed using PLINKv2.07.21
Patterns of MAF, LD and haplotype diversity, and comparison of the parameters in other populations
We estimated the MAF distribution, pairwise r2 and LD blocks for autosomal markers using PLINKv.2.07.21 The LD blocks were calculated as described23 over 500 kb segments. Further, we retrieved the mean length of LD blocks, the total number of blocks and the mean number of SNPs per block. To analyze haplotype diversity, we estimated the mean number of haplotypes per block.
To compare estimates of the extent of homozygosity, MAF and LD in Sardinians with other populations, we selected two other data sets for which genotypes derived from Affymetrix platforms were available, namely, the four main samples from the HAPMAP project and the POPRES database. The POPRES database contains the genotype matrix of over 4000 subjects classified by their country of origin based on PCA. This data set has been used to assess genetic structure in Europe and other continents.24, 25, 26, 27 Supplementary Tables S1a and S1b present the number of countries per population samples per data set. For PCA analysis within Europe, we analyzed all POPRES samples with proven evidence of European ancestry25 (Supplementary Table S1a). For analysis of LD and MAF distributions, we selected POPRES-European regions with at least 100 subjects. For POPRES-European regions with large samples, we selected a random subset of 200 individuals to avoid bias in the estimation of MAF and LD between the Sardinians and other samples from this data set (Supplementary Table 1Sb). Further, we estimated inbreeding, ROH segments, MAF, extent of LD and haplotype diversity in five POPRES-European regions, namely North-West Europe (Europe NW), Europe Central (Europe C), South Europe (Europe S), South-West Europe (Europe SW) and West Europe (Europe W). These samples had about 200 individuals each.
We tested for differences in the MAF distribution between Sardinians and other populations using Fisher's exact test using the R package v.2.10 (R Foundation for Statistical Computing, Vienna, Austria) based on allele counts and correcting for multiple testing using the Bonferroni correction. To compare the similarities of pairwise r2 and LD between the Sardinians and other populations, we calculated the Pearson correlation coefficients (r) over non-overlapping windows of 250 kb (with at least 50 r2 values). Further, we used Fisher's z-transformation to convert r values to a normally distributed z distribution28 and tested whether the Pearson correlation in a specific window was significantly lower than the genome-wide average on all autosomes. Differences in the length of blocks, number of blocks per autosome, mean number of alleles and haplotype diversity were tested using pairwise Mann–Whitney tests and correcting for multiple testing.
Results
Founder lineage heterogeneity
Table 1a presents the frequency distribution of the major haplogroups as defined by the UEPs based on 260 males. There were eight major haplogroups and 18 subclades, although only five of the latter accounted for 86% of the total haplogroup frequency distribution. The I2a1a haplogroup defined by the M26 mutation was the most prevalent, with a frequency of 42%. This haplogroup is absent in most European countries, attaining frequencies of 5% in the Basque population29 and 19% in Castilla (Spain).30 We also tested the M261 mutation that is derived from the I2a1a branch.31 We only identified three subjects with this mutation. Given the low frequency of this subclass, it is likely that it represents a recent mutation.
We compared the haplogroup frequencies in Sardinians with those reported in previous studies.15, 19, 32 First, we collapsed the subclades into major haplogroups, as not all the markers we used in our analysis were typed in previous publications. Table 2a presents the pairwise Fst as estimated between the Sardinian and other populations. As shown in previous studies,15, 19 Sardinia was clearly separated from nearby Mediterranean populations, with significant Fst estimates that varied from 9% (comparison with Greece) to 23% (comparison with Basques). We included the major Y chromosome haplogroup frequencies based on 373 Sardinian individuals described in Contu et al.15 We did not observe any significant differences between the two Sardinian samples, despite the fact that individuals were sampled from different regions15 (Table 2a). We also restricted the analysis to individuals from the ‘Barbagia’ region (Nuoro in Figure 1) and did not observe evidence for differentiation because of place of birth with the Y chromosome markers (data not shown).
We used the STR haplotypes to determine the extent of heterogeneity in the Sardinian sample and found a haplotype diversity of 98%. We compared the haplotype frequency distribution of six STR markers from which frequencies were available from other populations using Rst. We found significant Rst estimates, supporting the evidence for population differentiation, with similar genetic distances to those we found for the SNP haplogroups (data not shown).
Mitochondrial haplogroups were estimated. In our sample we could reliably determine haplogroups for 258 individuals. The haplogroup frequencies are presented in Table 1b. We observed 11 major clades with five of them accounting for 86% of all haplogroups. The most frequent major haplogroup was H, the most common European haplogroup, with a frequency of 46%. The next most frequent subclades were J2b, HV0 and U5b3a with frequencies of 5%, 5% and 4%, respectively. U5b3a appears to be specific to Sardinians, although the low frequency suggests that its origin is more recent than the Ia2a2 haplogroup of the Y chromosome.33
Mitochondrial haplogroup frequencies from the Sardinian sample were compared with the estimates from other populations.34, 35 Table 2b shows that, similar to the Y chromosome haplogroup system, there was population differentiation between the Sardinians and neighboring populations, but not with other samples from Sardinia. Estimates of gene diversity within the mitochondrial system were calculated for Sardinians and other populations. We found a gene diversity of 0.70 and mean heterozygosity of 0.6, which was much lower than the estimates obtained for the Y chromosome system.
Assessment of stratification, relatedness and homozygosity in the Sardinian population using a dense set of autosomal markers
Out of 113 individuals genotyped, we excluded six individuals from further analysis, including two duplicates, one recipient of a bone marrow transplant and three individuals that were born out of the sampling area. The mean genotyping call rate was 99.3% and 98.4% per markers and samples, respectively.
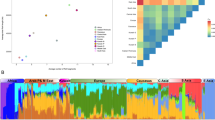
To investigate whether there was substructure in our sample we performed PCA. We identified nine outlier individuals who deviated from the rest by >6 SD and thus were excluded from further analysis. All nine individuals had genotype call rates below 98%. Figure 2 displays the plot of the two main axes derived from the PCA. In our analysis, we identified significant ‘eigen’ values, which suggested that there was stratification in the sample. However, this was not due to the fact that individuals were coming from different provinces (Figure 2), as we did not observe any cluster of individuals per region. Classifying the place of origin into Barbagia or the other regions did not reveal any clustering either (Supplementary Figure 1). We confirmed the absence of sub-population structure due to a differential genetic make-up, because the Fst estimates after grouping the provinces into Barbagia and ‘non-Barbagia’ regions were not significant.

PCA in Sardinians. Figure represents the first two components of PCA based on allele frequencies. Each point represents a subject and the six main sampling areas are depicted in different colors and shapes.
A close degree of relatedness among apparently unrelated individuals can lead to population structure.22, 36 We estimated the degree of relatedness in the Sardinian sample by calculating the average kinship coefficient between individual pairs. Out of 6323 pairs, we identified 980 (15%) that appeared to be closely related (π≥0.05). Of these, 74% had π estimates ≥0.0625 and <0.125 (<4 meiosis). None of the pairs were known to be related, consistent with non-random mating because of the past isolation of this population. Thus, in the Sardinian sample, the stratification we observed with PCA may be due to the high degree of relatedness among some individuals.
To scale the extent of genetic differentiation in Sardinians from other populations, we carried-out PCA analysis on allele frequencies from 99 534 autosomal markers in LE typed for the Sardinians as well as for the populations in the POPRES and HAPMAP databases. As shown in Supplementary Figure 2, the Sardinians formed a tight cluster with other European populations (POPRES and CEU samples). The pattern we observed for these samples is consistent with previous publications.37 We repeated the PCA including only European populations. Figure 3 shows that Sardinians clearly separate from other European populations. In addition, few individuals were scattered toward the Southern European sample (that included individuals from Italy). Of note these individuals were born in different villages, which rules-out the possibility that they form a separate cluster (genotype call rate in these individuals was above 98%). Sardinians were more similar to the POPRES samples than to the CEU sample. To gain insights into the differences and similarities of LD in Sardinians compared with other European samples, we used five samples from POPRES for the comparative analysis (see Subjects and methods), as these better reflect the actual population samples chosen for GWAS.

PCA in Sardinians, CEU and POPRES Europe. Figure represents the first two components of PCA based on allele frequencies. Each point represents a subject and regions from Europe are depicted in different colors and shapes.
Patterns of MAF
We analyzed the MAF distribution from 402 566 autosomal SNPs that passed QC. The median MAF was 0.18 (interquantile range: 0.06–0.34) and 8% of the SNPs were monomorphic. We compared the MAF distribution of SNPs in Sardinians with five European samples from POPRES based on the 318 665 SNPs common to all populations. The median MAF was 0.21 (IQ range; 0.08–0.35) for the POPRES-E samples. Figure 4 shows the MAF in Sardinians and POPRES-E. The SNPs that were monomorphic in the Sardinians were also rare or monomorphic in the European populations except for a small fraction (<1%) of SNPs that had frequencies between 5 and 10%. We tested for significant differences in the allele frequencies of every SNP between Sardinians and each of the European groups with Fisher's exact test. The proportion of SNPs with significant differences in allele frequency ranged from 5% for South and South-West European groups to up to 34% when Sardinians were compared with North-West European groups (Supplementary Figure 3).

MAF distributions of autosomal SNPs in Sardinians and five European regions.
Patterns of LD and haplotype diversity
We estimated pairwise correlations over 500 kb distances in Sardinians and six other European populations. Table 3 shows the average pairwise correlation per 250 kb window. The average correlation per chromosome was very similar across populations. This was reflected in the Pearson correlations between r2 measures. On average, r2 was 0.89 for the comparisons between Sardinians and Central Europeans and 0.91 for the comparisons between Sardinians and Southern Europeans. Similarly, in comparisons of MAFs between Sardinians and European populations, the populations that were more similar to Sardinians were from the South and South-West of Europe.
We tested for differences between the average genome-wide correlation in Sardinians and other populations, by looking for significantly lower Pearson correlations. Excluding telomeric regions, we found that 9% of 3894 windows had Pearson correlations that deviated significantly from the overall genome-wide estimate, with no significant differences in the comparisons between Sardinian-European pairs. Moreover, we identified 62 windows where the correlation in r2 estimates between Sardinians and other Europeans was ≤60%, and 34 of these regions harbored annotated genes (Hg 18 build) (Supplementary Table 2). Therefore, the LD structure of these genomic regions is different in Sardinians compared with other European populations. One region that stood-out with a lower correlation was the region encompassing the HLA-B system, as has been reported before.38
Table 4 presents the descriptive statistics of the LD blocks we derived for the Sardinian sample. There were three haplotypes per block, on average, which is similar to estimates obtained for other European populations.39 We compared the estimates with two population samples from POPRES: Europe North-West and Europe South-West. The number of LD blocks was larger for the two POPRES samples than for Sardinians. However, the mean number of haplotypes per block was similar to the estimates we obtained for the Sardinians. We also looked for actual differences in the LD block structure and haplotypes between Sardinians and the POPRES Europe North-West sample in the estimated LD blocks. Supplementary Figures 2 and 4 show that although the largest amount of blocks that were shared in both populations had identical haplotype structure, roughly 20% of blocks per chromosome were different, with an excess of rare haplotypes in both populations.
Extent of homozygosity in the genome of individuals from Sardinia and POPRES database
The extent of homozygosity in the Sardinian population was evaluated by estimating inbreeding using the set of SNPs in LE. In the Sardinian sample, 60 individuals out of 99 (58%) were inbred, and the median of coefficient values for these individuals was 0.01. This estimate was one order of magnitude larger than those we obtained from the European samples (Supplementary Table 3). The proportion of inbred individuals in the POPRES samples was also lower than the proportion of inbred subjects in Sardinia. Interestingly, the low inbreeding coefficients of Sardinians indicates that these individuals are homozygous because of consanguinity between their ancestors.
We estimated the median number of ROH segments per individual as well as the median length of homozygous segments. Overall, the average number of ROH segments per individual was similar across European populations (Supplementary Table 3). Sardinians deviated from the pattern of genome homozygosity we observed for POPRES. Indeed, as many as 80% of the Sardinian subjects had at least one ROH segment with a median of three segments per individual. The length of the ROH was also larger for the Sardinians. These analyses showed that Sardinians have an increased proportion of loci that are homozygous by descent, and that this is due to an excess of IBD sharing between ancestors of individuals in our sample.
Discussion
We characterized the genetic make-up of a sample from NE Sardinia using a dense set of autosomal, Y chromosome and mitochondrial markers. To our knowledge, we are the first to analyze both autosomal and uniparental markers in the same sample to address the extent of LD and gene heterogeneity in Sardinians as well as the degree of genetic substructure. We confirmed that Sardinians are genetically differentiated, because of genetic drift and past founder effects, as revealed by the high prevalence of the M26 mutation on the Y chromosome, the low frequency of a derived mutation of the Sardinian-specific Y chromosome haplogroup, as well as the moderate frequency of the UB53a mitochondrial haplogroup. The large number of founder lineages and the high haplotype diversity suggests either a large founding population size,40 or some degree of genetic admixture.
At the autosomal level we found, not surprisingly, that Sardinians were most similar to other Europeans, especially to individuals from South and South-West Europe, as patterns of pairwise LD and haplotype diversity were similar across these European populations. Nevertheless, we still found evidence for population differentiation between the Sardinians and other Europeans as revealed by PCA and MAF distributions. Indeed, PCA showed that Sardinians were a separate group when compared with other European populations (Figure 3b). We also found significant population differences in MAF between Sardinians and North-West and Central European populations (34% of MAF differences). In comparison, only 5% of MAF were different when Sardinian and South or South-West European populations were compared. This pattern resembles the observed gradient between genetic distance and geographical distances in European populations reported in other studies.26, 27 Our analysis of PCA and MAF distribution also shows that Sardinians are more similar to people from the South of Europe than to the populations of North-West and Central Europe including the CEU sample. This needs to be taken into account when designing genetic studies that include individuals from Sardinia.
Given the recent evidence of highly structured villages restricted to Ogliastra in Sardinia, we sought evidence for sub-population structure in our sample. We carried out PCA and found significant eigen vectors. However, we did not find any individuals that clustered based on their place of birth. Factors other than differential genetic background can lead to population stratification, including cryptic relatedness.36 In our sample, up to 15% of the individuals were related, which could partly explain structure in the sample. As missing data could also bias PCA results,22 we re-ran PCA with a higher threshold of missing data (5% for missing data on both samples and SNPs) and still found significant eigen values (data not shown). We also looked at the SNPs that correlated with the most significant eigen vectors (42 SNPs). The genotype call rate in these SNPs was 98% in average, and most of them were rare (MAF<5%), which might also lead to bias in PCA. Therefore, although we observed stratification in our sample, this was not due to individuals with different genetic backgrounds.
Our findings of genetic homogeneity in Sardinia contrast with those from a recent study where highly structured sub-population differentiation in a secluded area in Sardinia was shown.16 Pistis et al analyzed eight villages in the region of Ogliastra each with a unique demography, which are isolated from the rest of Sardinia and found significant Fst estimates between the villages, which was expected given the history of isolation of the regions. In contrast, the individuals from our study came from a much larger area comprising >80 villages from the NE of Sardinia. We did not find evidence for population differentiation using Y chromosome, autosome or mitochondrial markers. The difference between the two studies is most likely due to the difference in the sampling strategy. Our findings are in line with a previous report where there was no evidence of population differentiation across different regions in Sardinia.15 In fact, comparing our data with Contu et al, we did not find any significant Fst estimates although Contu et al sampled other regions. The frequencies of both Y chromosome and mitochondrial haplogroups is similar to these reported in other publications, which demonstrates that the sample is representative of Sardinia.
We identified genomic regions where the similarity of LD measures between Sardinians and other populations were significantly different. We observed that in 9% of genomic regions the Pearson correlation of the r2 estimates between Sardinians and other European populations was significantly lower and in some of these regions, including the HLA-B locus, the correlations were below 60%. This has important implications when designing GWAS using Sardinian samples, as LD structure might be different between control groups from different populations, leading to hidden stratification. This confirms the difference in local structure of LD between Sardinians and Europeans and shows that Sardinia is well suited for fine mapping of identified hits in GWAS studies in populations of European ancestry.
Sardinians still conserve signatures of their past isolation, including non-random mating and inbreeding. We observed increased genome homozygosity as revealed by the genome average inbreeding and ROHs that were significantly larger and more abundant in the genome of Sardinians when compared other European samples. These features might be useful when carrying-out a genome-wide analysis of ROH between cases and controls to identify traits in which dominant genetic variance (because of recessive alleles) is large. In addition, at some loci the increased MAFs because of founder effects could help to map disorders with increased prevalence in Sardinian, such as diabetes and multiple sclerosis.
To conclude, using a large set of autosomal, Y and mitochondrial markers, we show that Sardinians can be considered a genetically homogenous population with signatures of isolation and non-random mating that need to be considered when conducting GWAS. The genomes of individuals show that at least 9% of regions will differ in LD structure, irrespective of disease status, which is helpful to unravel functional variants using fine mapping. Moreover, the increased homozygosity in Sardinians is an advantage for mapping recessive variants by approaches such as shared haplotype analyses and genome-wide analysis of ROH.
References
Hindorff LA, Sethupathy P, Junkins HA et al: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106: 9362–9367.
Manolio TA, Collins FS, Cox NJ et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753.
Heutink P, Oostra BA : Gene finding in genetically isolated populations. Hum Mol Genet 2002; 11: 2507–2515.
Lowe JK, Maller JB, Pe’er I et al: Genome-wide association studies in an isolated founder population from the Pacific Island of Kosrae. PLoS Genet 2009; 5: e1000365.
Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS : The effect of genetic drift in a young genetically isolated population. Ann Hum Genet 2005; 69: 288–295.
Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV et al: Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet 2008; 40: 1402–1403.
Bonifati V, Rizzu P, van Baren MJ et al: Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science 2003; 299: 256–259.
Newman DL, Abney M, Dytch H, Parry R, McPeek MS, Ober C : Major loci influencing serum triglyceride levels on 2q14 and 9p21 localized by homozygosity-by-descent mapping in a large Hutterite pedigree. Hum Mol Genet 2003; 12: 137–144.
Caramelli D, Vernesi C, Sanna S et al: Genetic variation in prehistoric Sardinia. Hum Genet 2007; 122: 327–336.
Chio A, Borghero G, Pugliatti M et al: Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol 2011; 68: 594–598.
Service S, DeYoung J, Karayiorgou M et al: Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 2006; 38: 556–560.
Loudianos G, Dessi V, Lovicu M et al: Molecular characterization of Wilson disease in the Sardinian population—Evidence of a founder effect. Hum Mutation 1999; 14: 294–303.
Rosatelli MC, Dozy A, Faa V et al: Molecular characterization of beta-thalassemia in the Sardinian population. Am J Hum Genet 1992; 50: 422–426.
Sanna S, Pitzalis M, Zoledziewska M et al: Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet 2010; 42: 495–497.
Contu D, Morelli L, Santoni F, Foster JW, Francalacci P, Cucca F : Y-chromosome based evidence for pre-neolithic origin of the genetically homogeneous but diverse Sardinian population: inference for association scans. PLoS One 2008; 3: e1430.
Pistis G, Piras I, Pirastu N et al: High differentiation among eight villages in a secluded area of Sardinia revealed by genome-wide high density SNPs analysis. PLoS One 2009; 4: e4654.
Semino O, Passarino G, Oefner PJ et al: The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective. Science 2000; 290: 1155–1159.
Terwilliger JD, Hiekkalinna T : An utter refutation of the /‘Fundamental Theorem of the HapMap/’. Eur J Hum Genet 2006; 14: 426–437.
Francalacci P, Morelli L, Underhill PA et al: Peopling of three Mediterranean islands (Corsica, Sardinia, and Sicily) inferred by Y-chromosome biallelic variability. Am J Phys Anthropol 2003; 121: 270–279.
Excoffier L, Laval G, Schneider S : Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 2007; 1: 47–50.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Gabriel SB, Schaffner SF, Nguyen H et al: The structure of haplotype blocks in the human genome. Science 2002; 296: 2225–2229.
Auton A, Bryc K, Boyko AR et al: Global distribution of genomic diversity underscores rich complex history of continental human populations, 2009. Genome Res 19: 795–803.
Nelson MR, Bryc K, King KS, et al: The population reference sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet 2008; 83: 347–358.
Novembre J, Johnson T, Bryc K et al: Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Lao O, Lu TT, Nothnagel M et al: Correlation between genetic and geographic structure in Europe. Curr Biol 2008; 18: 1241–1248.
Sokal R, Rohlf F : Correlation; in WH Freeman and Co (ed.): Biometry. New York, 2003, Vol 1, pp 555–608.
Rootsi S, Kivisild T, Benuzzi G et al: Phylogeography of Y-chromosome Haplogroup I reveals distinct domains of prehistoric gene flow in Europe. Am J Hum Genet 2004; 75: 128–137.
Flores C, Maca-Meyer N, Gonzalez AM et al: Reduced genetic structure of the Iberian peninsula revealed by Y-chromosome analysis: implications for population demography. Eur J Hum Genet 2004; 12: 855–863.
Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF : New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 2008; 18: 830–838.
Di Gaetano C, Cerutti N, Crobu F, et al: Differential Greek and northern African migrations to Sicily are supported by genetic evidence from the Y chromosome. Eur J Hum Genet 2009; 17: 91–99.
Pala M, Achilli A, Olivieri A et al: Mitochondrial haplogroup U5b3: a distant echo of the epipaleolithic in Italy and the legacy of the early Sardinians. Am J Hum Genet 2009; 84: 814–821.
Fraumene C, Petretto E, Angius A, Pirastu M : Striking differentiation of sub-populations within a genetically homogeneous isolate (Ogliastra) in Sardinia as revealed by mtDNA analysis. Hum Genet 2003; 114: 1–10.
Torroni A, Richards M, Macaulay V et al: mtDNA haplogroups and frequency patterns in Europe. Am J Hum Genet 2000; 66: 1173–1177.
Price AL, Zaitlen NA, Reich D, Patterson N : New approaches to population stratification in genome-wide association studies. Nat Rev Genet 2010; 11: 459–463.
Nelson MR, Bryc K, King KS et al: The population reference sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am J Hum Genet 2008; 83: 347–358.
Fiorillo MT, Cauli A, Carcassi C et al: Two distinctive HLA haplotypes harbor the B27 alleles negatively or positively associated with ankylosing spondylitis in Sardinia: implications for disease pathogenesis. Arthritis Rheumatism 2003; 48: 1385–1389.
Pardo L, Bochdanovits Z, de Geus E et al: Global similarity with local differences in linkage disequilibrium between the Dutch and HapMap-CEU populations. Eur J Hum Genet 2009; 17: 802–810.
Thomas MG, Barnes I, Weale ME et al: New genetic evidence supports isolation and drift in the Ladin communities of the South Tyrolean Alps but not an ancient origin in the Middle East. Eur J Hum Genet 2008; 16: 124–134.
Acknowledgements
We thank all the participants of the study. We thank Dr Krishna R Veeramah for sharing software to format data into ARLEQUIN, and for his comments on the analysis of Y chromosome data. RA was supported by the ‘Master and Back’ program from the Regione Autonoma Sardegna. We thank Fabrizio Delussu for helpful discussions on the archeological and historical data from Sardinia. Statistical analyses were carried out on the Genetic Cluster Computer (GCC), GCC is financially supported by the Netherlands Scientific Organization (NWO 480-05-003). The POPRES data set was obtained from the dbGaP database through dbGaP accession no. phs000145.v2.p2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Pardo, L., Piras, G., Asproni, R. et al. Dissecting the genetic make-up of North-East Sardinia using a large set of haploid and autosomal markers. Eur J Hum Genet 20, 956–964 (2012). https://doi.org/10.1038/ejhg.2012.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.22
Keywords
This article is cited by
-
Mitochondrial DNA variability of the Polish population
European Journal of Human Genetics (2019)
-
Overcoming the dichotomy between open and isolated populations using genomic data from a large European dataset
Scientific Reports (2017)
-
Detecting past male-mediated expansions using the Y chromosome
Human Genetics (2017)
-
Genetic structure of the Newfoundland and Labrador population: founder effects modulate variability
European Journal of Human Genetics (2016)
-
Risk factors associated with colonic diverticulosis among patients from a defined geographic area
Techniques in Coloproctology (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}