
Abstract
Additional information about risk genes or risk pathways for diseases can be extracted from genome-wide association studies through analyses of groups of markers. The most commonly employed approaches involve combining individual marker data by adding the test statistics, or summing the logarithms of their P-values, and then using permutation testing to derive empirical P-values that allow for the statistical dependence of single-marker tests arising from linkage disequilibrium (LD). In the present study, we use simulated data to show that these approaches fail to reflect the structure of the sampling error, and the effect of this is to give undue weight to correlated markers. We show that the results obtained are internally inconsistent in the presence of strong LD, and are externally inconsistent with the results derived from multi-locus analysis. We also show that the results obtained from regression and multivariate Hotelling T2 (H-T2) testing, but not those obtained from permutations, are consistent with the theoretically expected distributions, and that the H-T2 test has greater power to detect gene-wide associations in real datasets. Finally, we show that while the results from permutation testing can be made to approximate those from regression and multivariate Hotelling T2 testing through aggressive LD pruning of markers, this comes at the cost of loss of information. We conclude that when conducting multi-locus analyses of sets of single-nucleotide polymorphisms, regression or multivariate Hotelling T2 testing, which give equivalent results, are preferable to the other more commonly applied approaches.
Similar content being viewed by others


Introduction
The emergence of genome-wide association (GWAS) technologies in recent years has resulted in the identification of over a thousand of novel risk loci for over 200 different complex phenotypes.1 While implementation of these technologies is now routine in a large number of laboratories, and individual loci occasionally can be confidently identified in small2 or moderate sized samples,3 in most instances, the effect sizes of the vast majority of common risk variants are small (OR<1.2) and their identification requires samples running into 10–100s of thousands.4, 5, 6 While studies on this scale are technically achievable, for many phenotypes there are insufficient samples available, and even where there are, the genotyping cost is a limiting factor. Thus, to date, there are few complex phenotypes where a high proportion of the genetic risk has been attributed to specific loci, even for those disorders where there is strong evidence that a substantial proportion of variation in liability is attributable to common single-nucleotide polymorphisms (SNPs).7, 8
The goal of uncovering the pathophysiology of complex disorders is best served by identifying the specific alleles involved, and characterizing their functional consequences at cellular and whole organism levels. However, this is not to say that useful information about disease pathophysiology cannot be attained through analytic approaches that fall short of identifying a comprehensive catalogueue of individual risk variants. Thus, it has been argued that useful insights into complex disorders can be provided by analyses in which evidence is accrued for association at the gene-wide level9, 10 or at the level of pathways derived from sets of genes11, 12 that are in some way functionally interconnected. Rather than focussing on individual SNPs, the analytic approach is based on extracting information from multiple SNPs within genes or within pathways and testing whether en masse, there is some significant excess of association signal in those genes or pathways. Such approaches may not be valid, or even required, for disorders where only a small number of genetic variants are involved. However, for those which can be considered essentially polygenic, involving hundreds or thousands of variants, it is intuitively likely that multiple independent risk factors will exist within related sets of genes, and even within individual genes.9, 10 Such a nonrandom dispersion of association signals among annotated gene sets has been empirically demonstrated in, for example, Crohn's disease and Bipolar disorder,11 and this has already been exploited to provide insights into the broad mechanisms underlying a number of phenotypes, including Alzheimer's disease13 and height.5 Moreover, the latter5 confirmed the hypothesis that a large number of loci have multiple independently associated variants. These findings support the hypothesis that gene-based multi-marker analyses might allow risk genes to be implicated where this cannot be achieved through analysis of single markers.
When considering multi-marker analyses, the best approach is still a matter of debate. The present study is concerned primarily with gene-wide analyses rather than multi-gene-based approaches where there are many more options available. We therefore restrict our analysis and discussion to approaches currently implemented for combining SNP data at the gene-wide level, although we note that they are also relevant when the same methods are applied to multiple genes.
In assessing the significance of a set of markers within a gene, it is necessary to consider not just the number of markers in the set and their P-values (or some other measure) but also the extent to which they are non-independent as a result of linkage disequilibrium (LD). Widely adopted approaches for assessing gene-wide significance are based on deriving either the product of all association P-values for SNPs in a gene (ProdP)10, 14, 15 or, alternatively, the average of their association χ2 statistics (AveTest; also denoted set-based analysis in PLINK16).
The ProdP test is based on Fisher's classical result that for M statistically independent random variables pj, which are uniformly distributed on the interval [0,1], the combined statistic 2Σj log(pj), j=1,…, M, follows a χ2 distribution with 2M degrees of freedom. By applying this principle to P-values obtained for a group of independent single markers, one can obtain an overall test of whether the distribution of P-values in the group of markers deviates from that expected by chance. In the presence of LD, the P-values are no longer independent, so Fisher's observation does not apply. To allow for this, significance is assessed empirically by randomly permuting case/control status, obtaining SNP P-values from the permuted datasets, combining these as in the observed dataset and by calculating the proportion of permutations in which the combined P-values are smaller than that in the observed data. The AveTest approach is based on the principle that if each single-marker test statistic is χ2 distributed with one degree of freedom, under the assumption of independence, their sum is χ2 distributed with M degrees of freedom. Again, LD is generally allowed for by permutation testing in a manner similar to that described for the ProdP. In theory, the ProdP and the AveTest are expected to give very similar results and this is the case in practice (see results). We should also note that Liu et al17 have recently suggested an approach using a simulated reference distribution rather than permutation of case–control status. This saves computation time and avoids the requirement for access to individual genotyping data, but as it gives essentially the same results as the ProdP and AveTest17 we do not discuss this method further.
The inspiration for the present study was an incidental observation that gene-wide P-values were highly sensitive to the degree to which included markers were pruned for pair-wise LD. Consider two scenarios, one in which two independent markers have been genotyped in a gene, the other where a third marker has been genotyped that is in perfect LD with one of the other two markers. As the addition of the third marker contributes no additional information (other than perhaps for quality control purposes), for a test to be valid, we would expect that the same results should be obtained under each scenario. However, in practice, the two scenarios frequently give very different results (see Results). Thus, we sought to compare the performance of these permutation-based approaches with other methods in the context of variable marker–marker LD.
Chapman and Whittaker18 have previously reported a comparative study of the power of several statistical tests for combining SNPs in a candidate region. For comparison with the permutation-based tests, we studied the Hotelling's T2 (H-T2) test,19 which Chapman and Whittaker18 also refer to as a ‘multivariate score test’ as it compares the differences between the multivariate means of two samples. When we denote by Xi the M-vector of genotypes (0, 1 or 2) at the marker loci for individual i, where i=1,…, n are in the case sample and i=n+1,…, n+m in the control sample, the H-T2 statistic is

where

is the vector of single-SNP scores and V−1 is the inverse of the corresponding marker–marker covariance matrix. This statistic follows a χ2 distribution with M degrees of freedom if V is known, or Hotelling's T2 distribution if V is estimated from the samples19 as

Note that the AveTest statistic corresponds to the situation where the off-diagonal elements of V are set to zero – that is, if LD is ignored. As well as having good power properties, the H-T2 has the additional advantages of being a classical test whose statistical properties are well understood and which are simple to implement. Chapman and Whittaker18 also explored a Bayesian score test20 as well as the method proposed by Wang and Elston.21 However, the former is not currently implemented for GWAS, and the latter did not perform well,18 so we did not investigate those approaches in the present study. Instead, we explored multi-marker logistic regression analysis.
Methods
Simulated data
To study the impact of extreme LD on the permutation-based methods, we simulated datasets of 500 cases and 500 controls in which genotypes at 10 SNPs were distributed under Hardy–Weinberg equilibrium with sampling assuming an underlying null hypothesis (ie, with equal allele frequencies in cases and controls). In the simulated datasets, minor allele frequencies (MAF) at each locus were 0.1 or 0.5 (1000 datasets for each MAF scenario). Two SNPs were independent while the other eight SNPs were all in perfect LD (r2=1) with each other and one of the two independent markers. For each simulated dataset, we performed 10 000 permutations to obtain empirical P-values using ProdP and AveTest.
We also assessed the performance of the H-T2 test and compared it with the ProdP test and regression analysis. As both H-T2 test and regression analysis exclude identical variables, we compared the performances of the methods for highly correlated SNPs (r2=0.95). For this we simulated 1000 datasets, under the assumption of no association, in which two SNPs were independent (MAFs=0.1 or 0.5) and the genotypes of the other eight SNPs were in strong LD (r2≈0.95) with one of the independent markers.
Observed GWAS data
For exploring the performance of the gene-wide tests in real data, we used a Bipolar dataset3 which consists of 1868 cases and 2938 controls typed with the GeneChip 500K Mapping Array Set (Affymetrix UK Ltd, HighWycombe, UK). The full details of the samples and methods for conduct of the GWAS are provided in the original study. Additional QC measures are described elsewhere.10 SNPs were assigned to genes if they were located within the genomic sequence between the start of the first and the end of the last exon of any transcript corresponding to that gene. The chromosome and locations for all SNPs and genes and their identifiers were taken from the human genome assembly build 36.2 of the National Center for Biotechnology Information (NCBI) database. In total, we retained 156 630 (41.5% of the total) SNPs, which mapped to 11 791 genes for which there was more than 1 annotated SNP (2–776 SNPs per gene).
Statistical analysis
For ProdP analysis, single-SNP association P-values were generated using the Armitage trend test (1 df). In the Bipolar dataset, the association test statistics for the markers we used are inflated above the null as estimated by the genomic control22 metric λ, which had the value λ=1.11. Although at least some of this inflation is likely attributable to the polygenic architecture of bipolar disorder;7, 8 all SNP association P-values were adjusted for λ. ProdP was calculated using an in-house C++ program. Hotelling tests and Logistic regression were performed using R-statistical software (www.r-project.org/) for simulated data. For the real data sets, Hotelling tests were calculated using an in-house C++ program adapted to process the binary PLINK formats.
To explore the impact of highly correlated markers in the real datasets, we LD pruned using PLINK, randomly removing one marker from pairs of correlated markers at a range of thresholds of r2.
Results
Permutation-based approaches using simulated datasets with highly correlated SNPs
First, we compared the permutation tests for 1000 simulated null datasets in which two SNPs in those datasets were independent (both MAF=0.1) and up to eight additional SNPs were in perfect LD with one of those markers. Figure 1a is a scatterplot of the empirical P-values obtained using ProdP for two independent markers alone compared with that obtained for three markers. In Figure 1b, we show the scatterplot comparing the set of the same two markers with 10 markers. Clearly, although they are correlated, the empirical P-values obtained when the set of SNPs includes a pair of identical markers (Figure 1a) are not identical to those when only the independent markers are included (mean difference in P-values=0.06, SD=0.047). The results show even greater divergence when the number of perfectly correlated markers is increased (Figure 1b; mean difference in P-values=0.15, SD=0.12).

ProdP analysis comparing 1000 simulated 2-marker datasets (MAFs=0.1 for both independent markers) with 3- (a) and 10-marker (b) datasets in which the additional markers are in perfect LD (r2=1) with one of the SNPs in the 2-marker set. 10 000 permutations were performed to estimate ProdP P-value for each simulated dataset. Each point represents an empirical P-value.
We repeated the same analyses for markers with MAF of 0.5 but the results were essentially identical (Supplementary Figure 2). Note that the discrepancies are not due to random effects inherent in permutation testing as precisely the same simulated data sets, and permutations thereof, were used to calculate the empirical P-values for the 2, 3 and 10 markers. As expected, analyses based upon the AveTest gave very similar results (Supplementary Figures 1 and 2) to the ProdP test and therefore for most of this manuscript, we report data based only on the ProdP method although the conclusions are applicable to the AveTest.
To illustrate how the discrepancies arise when including additional correlated markers, consider two independent markers with observed P-values p1=0.062 and p2=0.93 (Table 1, columns 2 and 3). The product of P-values corresponding to the two independent markers is 0.058 (the fourth column). The product of P-values of the two independent markers and eight copies of the second marker is 0.062 × 0.939=0.032 (the last column). Assume that these are the values that we obtain in our study. Performing only 10 permutations (Table 1), we obtain empirical P-values of 0.1 and 0.9 when we combine 2 and 10 markers, respectively (the row ‘Empirical P-values: Nperm=10’ of Table 1). Thus when permutations are performed for the set of SNPs (with eight additional copies of the second marker), the ProdP empirical P-value is large as the nonsignificant marker is given an unfair weight. Similarly, if the marker with multiple proxies happens to be nominally significant, then the significance of the set is likely to be inflated compared with the set of two independent markers. Note that this result does not simply reflect increased variance in the estimate of significance by permutation, the results obtained with 1000 and 10 000 permutations being essentially identical (Table 1). The sensitivity of AveTest/ProdP approaches to the numbers of SNPs in tight LD is an undesirable property as the inclusion of more than one such SNP adds no new information, it merely repeats the same association signal, and therefore should not alter the significance of the association test.
Comparison of permutation, H-T2 test and multi-marker logistic regression analysis
In our simulated data, as noted by Roeder et al,23 the H-T2 test and regression analysis gave essentially identical results (Supplementary Figure 4), and therefore we present results based upon only one of these approaches (H-T2) although we note regression has the additional advantage under some circumstances of being able to include study covariates.
To compare the properties of the H-T2 and permutation tests in a real dataset in which the distribution of LD may be more typical for a GWAS, we used the BD WTCCC dataset.3 As our analysis is predicated on multiple SNPs per gene, we excluded genes represented by only one SNP in the dataset, retaining 11 791 genes. As can be seen in Figure 2, the disagreement between the P-values obtained from ProdP test (x-axis) and H-T2 test (y-axis) is profound (correlation=0.61, mean of differences for P-values=0.19, SD=0.18).

H-T2 test versus ProdP gene-wise P-values in the WTCCC BD dataset (11791 genes): (a) in nominal scale, (b) in logarithmic scale.
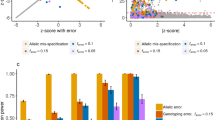
Given the results in the simulated data, one likely reason for the marked discrepancy between the results of the ProdP and the H-T2 tests is the presence of LD. The impact of this is illustrated in Figure 3. In Figure 3a, each point on the graph represents the value of the Z-statistics for two independent SNPs derived from 1000 simulated datasets of 500 cases and 500 controls under the null hypothesis. The assumed MAF here is 0.5 but similar results are obtained for other MAFs. The circle outlines the critical values corresponding to the 5% significance level, the data following a standard two-dimensional normal distribution. For pairs of markers outside the circle, the AveTest result (and ProdP) for the two SNPs will be nominally significant (we refer here first to the AveTest result rather than ProdP because it is simpler to plot when the data are represented by Z-scores). Under the scenario of two independent SNPs, the AveTest/ProdP and H-T2 tests give essentially identical results (red points overlap with blue circles). In Figure 3b, we plot a similar set of scenarios but the markers are no longer independent (r2=0.8), and hence the Z-scores for pairs of markers are correlated. As before, the theoretical boundary corresponding to the 5% significance level is plotted. Based upon probability theory, this is described by an ellipse whose boundaries and shape are defined by the degree of correlation between the two markers. It can be seen that the H-T2 test correctly specifies those beyond the ellipse as nominally significant and those within the ellipse as not significant (P≤0.05). However, the boundary corresponding to 5% significance for the AveTest/ProdP is still the dashed circle.

Multivariate sampling distribution for two independent markers (a) and two markers in r2=0.8 (b) for 1000 simulated datasets of two SNPs with MAF=0.5. The circle and ellipse indicate the significance threshold at α=0.05 for the joint distribution of two standard normally distributed variables. Samples flagged as significant (α=0.05) by ProdP and H-T2 are indicated by blue circles and red points, respectively.
Regardless of the correlation between a pair of SNPs, in set-based analysis (which according to theory, and which we show empirically, is equivalent to ProdP) the test statistic from which the significance level is derived is the average of the χ2s. For each single-SNP test, the χ2 is equal to the square of the z-score. Therefore the test statistic T=0.5·(Z12+Z22), where Z1 and Z2 are the z-scores for the two SNPs, or 2T=Z12+Z22. It is well established that the equation r2=x2+y2 denotes a circle of radius (r) in the (x,y) plane (ie, a standard plot with axes x and y) whose circumference passes through all values of x and y for which the √(x2+y2) are equal to the radius. From 2T=Z12+Z22, it follows that we also have a circle in the (Z1, Z2) plane whose radius=√2T. It is therefore a simple matter to plot any given circle whose radius is defined by the average χ2 corresponding for any given type-I error rate.
Note that the type-I error rates for H-T2 and AveTest/ProdP are both the same, but that the AveTest/ProdP disagrees with the H-T2 by designating moderate associations arising from correlated markers as significant while overlooking stronger effects of less-correlated markers. Both types of discrepancy occur with equal frequency, since the type-I error rate is unchanged. These instances appear in Figure 3b as empty blue circles, denoting scenarios where the ProdP test wrongly (benchmarked against probability theory) accepts or rejects the null. In contrast, the rejection regions of the H-T2 test correspond to those defined by Equation (1). Given that the addition of several SNPs in tight LD (essentially, repeating the same signal) will have a large effect on the significance of AveTest/ProdP, but not H-T2, we conclude that the H-T2 test is preferable to the AveTest/ProdP approaches in the context of LD. It should be noted that the GWAS in which we compared H-T2 and AveTest/ProdP approaches (Figure 2) was performed based on what would now be considered a low-density chip, and so that analysis will underestimate the impact of residual marker–marker LD on the ProdP test.
Given that the ProdP test thus has systematic disadvantages in the context of LD, we next investigated at what point the results of H-T2 and ProdP tests become similar when the strength of correlation between markers is reduced. Figure 4a presents the data for r2=0.8 and Figure 4b for r2=0.05. As expected, randomly pruning correlated SNPs in genes at increasingly stringent LD thresholds (r2=0.8, 0.5 and 0.05) resulted in a convergence of the results from ProdP and H-T2. The correlations between the P-values are 0.71 and 0.98 for (A) and (B), respectively, compared with 0.61 for the unpruned data. Note that to make a fair comparison between the analyses based upon different thresholds for pruning, analyses are restricted to those genes (N=7933) that had more than one SNP after pruning at the most stringent threshold r2=0.05.

H-T2 versus ProdP gene-wise P-values in the WTCCC BD dataset. SNPs were randomly pruned to reduce LD within the datasets at r2=0.8 (a) and r2=0.05 (b). The same SNPs are included in the H-T2 and ProdP analyses.
While pruning improves the concordance between the tests, an important question arises whether by removing correlated SNPs we also remove important information. This could occur for two reasons. First, under a random pruning procedure, the most significant signal in any gene may not be retained. However, this can easily be avoided by retaining the most significant marker from pairs in LD (and also doing so in the permutation process). Less trivially, the main reason to undertake gene-wide tests is to exploit the existence of multiple sources of association signals within a gene,9, 10 and it may well be that distinct functional variants are not fully independent (ie, within a gene, two distinct functional variants may be in partial LD). Similarly, even if there is only a single functional variant, multiple SNPs that are partially correlated may capture more of the association signal than does a single SNP. In order to explore the possibility of loss of power to detect association by removing SNPs in high LD, we compared the results obtained from analysis of the WTCCC BD dataset for pruned and unpruned data. In Figure 5a, the coordinates of each point on the scatterplot are the P-values for a gene where SNPs were LD-pruned data (x-axis) and unpruned (y-axis). If there is a loss of power, there will be a reduction in the number of points above the y=x line (Figure 5a) and in the QQ plot (Figure 5b); should a loss of power occur, the curve will deviate (compared with unpruned) towards the diagonal (ie, the null).

Gene-wise P-values by H-T2 test comparing unpruned genes against randomly pruned genes with r2=0.05 (a) and 0.8 (b).
Thus, Figure 5 demonstrates that when the H-T2 is applied, even modest random pruning (r2=0.8) tends to decreases the gene-wise H-T2 statistics, from which we infer loss of power. Deviation of the curve towards the null is greater with more rigorous pruning, such that at r2=0.05, the degree of pruning at which ProdP and H-T2 tests become comparable (Figure 4), the curve of observed gene-wide P-values has deviated dramatically towards the null.
To examine whether use of the truncated ProdP14 (TProdP) method improved concordance with H-T2, we ran 1000 permutations of the BD dataset using TProdP to calculate gene-wide P-values using truncation thresholds for individual SNPs for P-values of 1 (the same as ProdP), 0.05, 0.01 and 0.001. Unsurprisingly, truncation resulted in substantial reductions in the number of genes in the analysis as many genes do not contain SNPs with P-values smaller than the truncation thresholds. Thus, after removing SNPs with P-values greater than or equal to 0.05, 0.01 and 0.001, respectively, only 2811, 813 and 109 genes remained (out of 11791 genes with more than 1 SNP), see Supplementary Table 1. We compare the gene-wide P-values for the TProdP and H-T2 tests in Supplementary Figures 5 and 6. The ProdP and TProdP results are discrepant but quite correlated (Supplementary Figure 5), correlations range between 0.31 and 0.9 (Supplementary Table 2). The discrepancy is more pronounced when TProdP is compared with H-T2 (Supplementary Figure 6), with the correlations between the gene-wide P-values calculated using TProdP and H-T2 being a maximum of 0.35 (see Supplementary Table 2). Note that the truncation procedure also excludes large numbers of genes that are significant through H-T2 analysis (Supplementary Table 1). For example, after truncation at P=0.05, only 29 genes are considered significant at P<0.001 compared with 134 of the full set of genes by H-T2. Interestingly, Supplementary Table 2 shows that, in addition to reducing the number of genes for which gene-wide P-values can be calculated, truncation also appears to reduce the correlation between ProdP and H-T2 P-values.
Conclusions
In this paper, we compare the performance under the null hypothesis of four different statistical tests for association in case/control data for a number of sets of genetic markers. They all combine, in different ways, the results of single-marker association for the group of markers under consideration with the aim being to generate an overall single-association test for the group. The ProdP test and the AveTest gives virtually identical results, as do the H-T2 test and logistic regression; these form two distinct groups of tests for which we have chosen ProdP and H-T2 as representatives.
We have shown that despite the use of permutations, the results of the ProdP (and AveTest) approaches are sensitive to the inclusion of highly correlated markers. This sensitivity arises from the relatively high weighting given to sets of markers that are correlated with additional typed variants over those that are not. Given that such markers are essentially repeating the same association signal, and therefore should not affect the significance of the association test, this sensitivity must be regarded as an undesirable property. We also show that in the case of a GWAS dataset, which includes highly correlated markers, the results of H-T2 and ProdP tests are very different.
Although the permutation-based tests are adjusted to preserve the type-I error, they differ from H-T2 test in the character of their type-II errors. By overweighting the impact of highly correlated markers, the ProdP test might allow the detection of associations where the data are driven by highly correlated SNPs, which are individually significant (empty blue circles), but this is at the cost of missing the joint independent effects of SNPs with small (non-significant) effects (the red dots without blue circles in Figure 3). We simulated several scenarios of a number of tightly correlated SNPs and one uncorrelated SNP (see Supplementary Material for details) under the null hypothesis of no association at any SNP, when one of the block of SNPs is a disease-susceptibility SNP and finally when the uncorrelated SNP is a disease-susceptibility SNP. Both tests give the same type-I error, the power of H-T2 test is higher when one uncorrelated SNP is associated with the disease and is lower when the associated SNP is one of the correlated SNPs in the block. However, significant associations driven by individually significant markers can be identified by single-marker analysis, and the question is how often in practice do the patterns of association favour the ProdP versus the H-T2. In the WTCCC BD dataset (Figure 2 and Supplementary Table 2), at the 5, 1 and 0.1% significance thresholds, the percentage of genes surpassing these thresholds using the H-T2 test are, respectively, 12.6% (N=1480), 4% (N=505) and 1.14% (N=134) compared with 5.97% (N=704), 1.57% (N=185) and 0.30% (N=35) for ProdP, from which we can infer that ProdP has lower power than H-T2. Thus, even ignoring its better theoretical performance, the H-T2 test also has greater power in a real dataset. We have also observed a similar superior performance for the H-T2 in other GWAS datasets (data not shown).
The results of ProdP analysis can be forced to converge with those expected from theory (Figure 3) and from H-T2 analysis (Figure 4) as markers become less dependent through more aggressive LD pruning. However, the r2 threshold for pruning at which this occurs is quite low, r2=0.05 (Figure 4b), and as shown in Figure 5, when markers are LD pruned, changes to the scatter and QQ plots suggest considerable loss of power (Figure 5b).
In conclusion, we have shown that for multi-markers analysis the results obtained using the ProdP test are not robust to the inclusion of highly correlated markers, the impact of which are given excessive weighting. We also show that the ProdP and H-T2 tests give remarkably divergent results, and that in the context of correlated markers, the H-T2 but not the ProdP performs well in comparison with probability theory. Although the results from the two tests can be forced to converge through aggressive LD pruning, that process reduces the information captured by, and the power of, the multi-locus tests. In unpruned data, the power of the H-T2 and ProdP methods vary with the patterns of LD around disease-susceptibility variants. Although in principle, the ProdP test may afford advantages if a high proportion of the additional association signal that can be accessed through gene-wide tests comes from very highly correlated significant markers, in real datasets, the H-T2 test appears more powerful. We conclude therefore that H-T2 test (and equivalently regression-based approaches) is preferred over the ProdP given both the superior validity in the light of probability theory, and its empirical superior performance in real datasets.
References
Hindorff LA, Junkins HA, Hall PN, Mehta JP, Manolio TA : A Catalog of Published Genome-Wide Association Studies. www.genome.gov/gwastudies (accessed April 2011).
Klein RJ, Zeiss C, Chew EY et al: Complement factor H polymorphism in age-related macular degeneration. Science 2005; 15: 385–389.
Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447: 661–678.
Speliotes EK, Willer CJ, Berndt SI et al: Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010; 42: 937–948.
Lango Allen H, Estrada K, Lettre G et al: Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010; 467: 832–838.
Voight BF, Scott LJ, Steinthorsdottir V et al: Corrigendum: twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2011; 43: 388.
International Schizophrenia Consortium, Purcell SM, Wray NR et al: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Lee SH, Wray NR, Goddard ME, Visscher PM : Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 2011; 88: 294–305.
Neale BM, Sham PC : The future of association studies: gene-based analysis and replication. Am J Hum Genet 2004; 75: 353–362.
Moskvina V, Craddock N, Holmans P et al: Gene-wide analyses of genome-wide association datasets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psych 2009; 14: 252–260.
Holmans P, Green EK, Pahwa JS et al: Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 2009; 85: 13–24.
Wang K, Li M, Bucan M : Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 2007; 81: 1278–1283.
Jones L, Holmans PA, Hamshere ML et al: Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS ONE 2010; 5: e13950.
Zaykin DV, Zhivotovsky LA, Westfall PH, Weir BS : Truncated product method for combining P-values. Genet Epidemiol 2002; 22: 170–185.
Dudbridge F, Koeleman BPC : Rank truncated product of P values, with application to genomewide association scans. Genet Epidemiol 2003; 25: 360–366.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet 2007; 81: 559–575.
Liu JZ, Mcrae AF, Nyholt DR et al: A versatile gene-based test for genome-wide association studies. Am J Hum Genet 2010; 87: 139–145.
Chapman J, Whittaker J : Analysis of multiple SNPs in a candidate gene or region. Genet Epidemiol 2008; 32: 560–566.
Hotelling H : The generalization of Student's ratio. Ann Math Stat 1931; 3: 360–378.
Goeman JJ, van de Geer SA, van Houwelingen HC : Testing against a high-dimensional alternative. J R Stat Soc 2005; 68: 477–493.
Wang T, Elston RC : Improved power by use of a weighted score test for linkage disequilibrium mapping. Am J Hum Genet 2007; 80: 353–360.
Devlin B, Roeder K : Genomic control for association studies. Biometrics 1999; 55: 997–1004.
Roeder K, Bacanu SA, Sonpar V, Zhang X, Devlin B : Analysis of single-locus tests to detect gene/disease associations. Genet Epidemiol 2005; 28: 207–219.
Acknowledgements
This study makes use of data generated by the Wellcome Trust Case Control Consortium (www.wtccc.org.uk) and the International Schizophrenia Consortium. This research was supported by grants from the MRC (UK) and by a NIMH (USA) CONTE: 2 P50 MH066392-05A1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Moskvina, V., Schmidt, K., Vedernikov, A. et al. Permutation-based approaches do not adequately allow for linkage disequilibrium in gene-wide multi-locus association analysis. Eur J Hum Genet 20, 890–896 (2012). https://doi.org/10.1038/ejhg.2012.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.8
