
Abstract
The analytical algorithm of Lynch syndrome (LS) is increasingly complex. BRAF V600E mutation and MLH1 promoter hypermethylation have been proposed as a screening tool for the identification of LS. The aim of this study was to assess the clinical usefulness and cost-effectiveness of both somatic alterations to improve the yield of the diagnostic algorithm of LS. A total of 122 colorectal tumors from individuals with family history of colorectal cancer that showed microsatellite instability and/or loss of mismatch repair (MMR) protein expression were studied. MMR germline mutations were detected in 57 cases (40 MLH1, 15 MSH2 and 2 MSH6). BRAF V600E mutation was assessed by single-nucleotide primer extension. MLH1 promoter hypermethylation was assessed by methylation-specific multiplex ligation-dependent probe amplification in a subset of 71 cases with loss of MLH1 protein. A decision model was developed to estimate the incremental costs of alternative case-finding methods for detecting MLH1 mutation carriers. One-way sensitivity analysis was performed to assess robustness of estimations. Sensitivity of the absence of BRAF mutations for depiction of LS patients was 96% (23/24) and specificity was 28% (13/47). Specificity of MLH1 promoter hypermethylation for depiction of sporadic tumors was 66% (31/47) and sensitivity of 96% (23/24). The cost per additional mutation detected when using hypermethylation analysis was lower when compared with BRAF study and germinal MLH1 mutation study. Somatic hypermethylation of MLH1 is an accurate and cost-effective pre-screening method in the selection of patients that are candidates for MLH1 germline analysis when LS is suspected and MLH1 protein expression is absent.
Similar content being viewed by others

Introduction
Lynch syndrome (LS) is characterized by an autosomal dominant inheritance of early-onset colorectal cancer (CRC) associated with an increased risk of other cancers.1, 2 It is caused by germline mutations in DNA mismatch repair (MMR) genes being MLH1 and MSH2 the most commonly mutated.3, 4, 5 Genetic heterogeneity and the low prevalence of hereditary tumors make it expensive to test all patients in whom LS is suspected.
Microsatellite instability (MSI) is a hallmark of MMR-deficient cancers and is found in >90% of LS colorectal tumors.6, 7 Immunohistochemistry staining is also used to determine the loss of expression of MMR proteins in tumor tissue of candidate patients. In spite of a low sensitivity, both strategies are generally accepted as prescreening procedures for genetic testing of MMR genes.8, 9
BRAF V600E mutation is present in approximately 10% of CRCs and in a higher proportion of MSI tumors. This mutation is strongly associated with MLH1 inactivation secondary to promoter hypermethylation.10, 11, 12, 13, 14, 15 It has been used to distinguish LS-associated from sporadic MSI-positive tumors.10, 11, 16, 17, 18, 19, 20, 21 The lack of BRAF mutations identifies with high sensitivity (96–100%) and lower specificity (22–100%) CRC cases associated with LS.10, 11, 16, 17, 18, 19, 20, 21 Occasionally, BRAF mutations have been detected in LS patients.22
Methylation of the MLH1 promoter, leading to a loss of MLH1 expression, is also strongly associated with sporadic MSI-positive CRCs. MLH1 promoter hypermethylation has been also evaluated for the selection of patients that will not be tested for germline mutation.23, 24, 25 However, the identification of hypermethylation in a limited number of LS tumors has made its use controversial.10, 26, 27, 28
Issues that affect screening include the accuracy, sensitivity, and specificity of the test, the benefit to the patient, the possible negative ramifications of the results, and the cost.8, 29, 30, 31 Before routine implementation in the clinical setting, it is critical to assess the analytical and clinical validity and the cost-effectiveness of BRAF mutation and MLH1 promoter hypermethylation.
The aim of this study was to compare the diagnostic yield and cost-effectiveness of BRAF V600E mutation versus MLH1 promoter hypermethylation in a large series of cases with familial aggregation for which MMR gene status was studied.
Materials and methods
Samples and patients
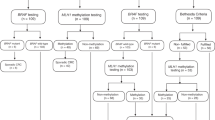
A total of 122 colorectal tumors with MMR deficiency (as evidenced by MSI or combined MSI and loss of MMR protein expression) were obtained from individuals with family history of CRC attended at our Cancer Genetic Counseling Unit between 1999 and 2008. A total of 43 patients met Amsterdam criteria, 48 revised Bethesda criteria and 12 cases showed other types of CRC familial aggregation. In all cases, MMR germline mutation status was assessed by direct sequencing and multiplex ligation-dependent probe amplification (MLPA). Overall, 57 tumors were from LS patients (40, 15 and 2 with MLH1, MSH2 and MSH6 mutations, respectively). In addition, a series of 48 (MSS) tumors from patients showing CRC familial aggregation and 73 sporadic CRC from a case–control study 32 were also analyzed. Informed consent was obtained from all patients, and the ethics committee approved this study.
DNA extraction from formalin-fixed paraffin-embedded (FFPE) material was done after microdissection of tumor cells using the QIAmp DNA Mini Kit (Qiagen, Hilden, Germany). DNA from RKO colorectal tumor cell line (ATCC, Manassas, VA, USA) was used as a biallelic MLH1 methylation control. DNA from SK-MEL-28 melanoma cell line (ATCC) and from COLO 201 colorectal cell line (kindly provided by Dr Soong) were used as controls of homozygous BRAF V600E mutation. To generate unmethylated DNA, peripheral blood lymphocyte (PBL) DNA was amplified using the REPLI-g kit (Qiagen).
Detection of somatic alterations
Detection of BRAF V600E mutation
A 196-bp region of exon 15 of the human BRAF spanning the hotspot mutation c.1799T>A. (V600E) was amplified by PCR and subsequently sequenced (as described in Supplementary Data Table 1). BRAF V600E mutation detection was also performed by Single Nucleotide Primer Extension (SNuPE) using the ABI PRISM SNaPshot Multiplex Kit (Applied Biosystems, Foster City, CA, USA) with specific primers (as described in Supplementary Data Table 1). Analytical sensitivity of BRAF V600E mutation analysis was assessed in serial dilutions of homozygous V600E mutated DNA from SK-MEL-28 and COLO 201 cell lines with wild-type genomic DNA from PBL.
Detection of MLH1 promoter methylation status
Methylation-specific MLPA (MS-MLPA) SALSA MS-MLPA ME011 kit (MRC Holland, Amsterdam, The Netherlands) is based on the use of probes that contain a digestion site (or occasionally two digestion sites) specific for the methylation-sensitive HhaI enzyme. Five pair of probes target A to D regions in MLH1 promoter and intron 1 (Figure 1). Analytical sensitivity was assessed in serial dilutions of RKO DNA and unmethylated DNA. Intra- and inter-experiment variability was assessed using a tumor sample showing methylation values close to 20%. A similar value has been proposed as a meaningful cutoff value in previous studies.33, 34 Ten replicates in two independent experiments were analyzed.

Detailed methylation patterns of MLH1 gene promoter, as assessed by MS-MLPA, of the 71 familial CRC tumors showing loss of MLH1 protein expression. The five regions of the CpGs targeted by the selected probes are shown. Samples lacking or harboring germline MLH1 gene mutations are separately described in panels a and b, respectively. Box highlights the methylation pattern of the informative C and D regions.
Methylation-specific melting curve analysis (MS-MCA) MS-MCA method consists of a real-time PCR followed by temperature dissociation35 on DNA previously treated with sodium bisulfite, using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA). For experimental conditions and primer sequences see Supplementary Data Table 1. Analytical sensitivity of the method was assessed as described above.
Pyrosequencing Two μl of bisulfite converted DNA were used in a PCR reaction of the regions of interest using HotStar Taq master mix (Qiagen), and biotin-labeled primers. Primers were designed using the Pyromark Assay Design Software 2.0 (Qiagen). For experimental conditions and primer sequences see Supplementary Data Table 1.
Analysis of MLH1 loss of heterozygosity (LOH) in the MLH1 mutation carrier
Allelic imbalances and copy number variation were analyzed using SNuPE technique and MLPA, respectively. Experimental conditions are described in Supplementary Data Table 1.
Cost-effectiveness analysis
A decision model was developed to estimate the number of mutation carriers and the incremental costs of alternative case-finding methods for detecting MLH1 mutation carriers among individuals with a positive molecular test in tumors (MSI and/or loss of expression of MLH1). Strategy 1 (BRAF – MLH1 mutation analysis) involved BRAF V600E testing of all individuals. If no mutation was detected, MLH1 mutation testing followed. Strategy 2 (Hypermethylation – MLH1 mutation analysis) involved testing for MLH1 hypermethylation of all individuals. If hypermethylation was absent, MLH1 mutation testing followed. Strategy 3 (MLH1 mutation analysis) involved direct MLH1 mutation testing of all individuals.
Pathway probabilities were attached to the decision tree (Table 1 and Supplementary Data Figure 1). For each strategy, the number of individuals tested, mutations detected and missed, false-positive results, and MMR mutations detected in first- and second-degree relatives were computed. Resource valuations were attached to each event in order to calculate a total cost for each strategy (Supplementary Data Table 2). The health outcome was defined in terms of additional MMR mutations detected. Two independent assessments were performed: one that considered costs and outcomes for proband only, and a second that included proband and their first- and second-degree relatives. Following a healthcare perspective, direct medical costs were used for analysis.
Finally, a one-way sensitivity analysis was performed in order to evaluate the relative impact of distinct parameters on the incremental cost per additional MLH1 mutation carrier detected. Each parameter is individually tested over a range of values while holding all other parameters at their base-case values. Ranges for each variable were based on a literature review or on expert opinion if no range data were available.
Results
Usefulness of BRAF V600E mutation analysis in the identification of LS tumors
SNaPshot consistently detected the BRAF V600E mutation when it was present in 5% of all alleles analyzed (Supplementary Data Figure 2). BRAF mutation was identified in 5 of 24 (20%) MSI and 2 of 49 (4%) MSS sporadic tumors. BRAF mutations were occasionally detected in MSS tumors of cases with familial aggregation.
BRAF mutations were detected in 14 of 122 (11%) MSI tumors. One BRAF mutation was identified in a tumor from a patient with MLH1 germline mutation. Absence of BRAF mutations was highly sensitive (98%; 56/57) for the identification of LS tumors. Specificity was 20% (13/65). All cases harboring BRAF V600E mutations were associated with loss of MLH1 expression. Restricting the analysis to the 71 tumors with loss of MLH1 protein expression (24 LS and 47 non-LS tumors), the absence of the mutation showed a sensitivity of 96% (23/24) and a specificity of 28% (13/47) for depiction of LS tumors (Supplementary Data Table 3).
Usefulness of MLH1 promoter methylation analysis in the identification of LS tumors
Following Deng et al36, only positive results for C- and D-regions (Figure 1) were scored as hypermethylation, as both correlate with loss of expression. MS-MLPA analytical sensitivity was 10% (Figure 2 and data not shown). Intra- and inter-experiment variability of MS-MLPA was within the range of 1%.

Analytical sensitivity and experimental variability of MLH1 promoter hypermethylation analysis as assessed by MS-MLPA. (a) Its performance was tested by serial reconstitutions of methylated alleles in increasing amounts of unmethylated alleles ranging from 100 to 0%. A linear relationship was detected between observed and predicted methylation. (b) Intra- and inter-experimental variability values refer to those obtained in a borderline sample showing methylation close to 20%.
The reconstituted samples were also analyzed using pyrosequencing. Its analytical sensitivity was 5% for C-region and 10% for D-region (Supplementary Data Table 4) similar to that of MS-MLPA (Supplementary Data Figure 3). Of note, the intensity of the methylation signal of MS-MLPA-targeted CpGs is average compared with the methylation for the whole region as assessed by pyrosequencing.
In the set of 71 tumors with MLH1 loss, MS-MLPA analysis evidenced MLH1 promoter hypermethylation in 32 cases when a cutoff value of 20% was used (Supplementary Data Table 3). Absence of hypermethylation showed a sensitivity of 96% (23 of 24) and a specificity of 66% (31 of 47) for LS identification. Two cases displayed exclusive MLH1 C-region methylation and three tumors displayed exclusive D-region methylation (Figure 1). If we would have considered methylation in regions A or B, six additional cases would have been misclassified as false-negative cases (Figure 1). Finally, the combination of BRAF mutation and MLH1 hypermethylation did not yield any additional value (Supplementary Data Table 3).
In our experience, the 20% cutoff value for MS-MLPA for MLH1 promoter assessment proved to be useful. However, the use of distinct cutoff values affects its putative clinical usefulness (Supplementary Data Table 3). The 20% cutoff usefulness was validated in an additional set of 10 cases (4 LS and 6 non LS), where it adequately classified all cases (data not shown). Alternative methods to assess methylation status were also evaluated. A MS-MCA test was developed that showed an analytical sensitivity of 5% (Supplementary Data Figure 3). Subsequently, a set of six tumor samples for which methylation levels were estimated between 5 and 20% by MS-MLPA were analyzed. MS-MCA scored as methylated one case previously scored as unmethylated. However, MS-MCA failed to identify as methylated a case showing 20% methylation levels (data not shown). MS-MCA did not add value in those cases with borderline values according to MS-MLPA.
In all, only one LS-associated colorectal tumor harbored somatic MLH1 promoter hypermethylation and a BRAF mutation (Supplementary Data Figure 4). This tumor arose in a patient that fulfilled Bethesda criteria. His first tumor was located in the right colon and diagnosed at 23-years old. The second was diagnosed at age 43 and was located in the sigma (pT3pN2M0). The latter tumor was the one analyzed. Family history included a diagnosis of CRC of his mother, whereas his father developed a gastric cancer. Tumor tissue study revealed neither somatic copy number variation of the MLH1 gene nor LOH was evidenced. Although the tumor displayed the typical molecular profile associated with sporadic MSI tumors, BRAF mutation and MLH1 promoter hypermethylation, the patient was a carrier of the founder Spanish pathogenic MLH1 c.1865T>A (L622H) mutation.37
Cost-effectiveness analysis
Both testing strategies were compared with MLH1 germline testing for all individuals (BRAF–MLH1 mutation analysis (Strategy 1); Hypermethylation - MLH1 mutation analysis (Strategy 2); DNA testing of all individuals (Strategy 3) (Supplementary Data Figure 1). Parameters, base case values and assumptions used to calculate the incremental cost per additional mutation detected are shown in Table 1.
The three strategies were analyzed in a hypothetical cohort of 1000 newly diagnosed CRC patients with loss of MLH1 expression (Table 2). DNA testing of all probands (Strategy 3) is anticipated to identify all expected carriers (n=338) (Table 2). Strategies 1 and 2 identified the same number of carriers (n=324), but strategy 1 associated with a higher number of false-positive when compared with strategy 2 (479 vs 165). When first- and second-degree relatives were also considered, the number of identified MLH1 mutation carriers increased up to 810, improving the clinical impact of the screening. Strategy 3 was able to identify 35 additional cases (Table 2).
Strategy 2 offered the lowest cost per additional mutation detected (Table 2). When probands were considered, the incremental cost for the identification of an additional MLH1 mutation was 2212 for strategy 2, whereas for strategy 3, the most specific one, the incremental cost was 27 220 (Table 2). When costs and benefits were calculated including first- and second-degree relatives, the incremental cost per additional mutation detected for strategies 2 and 3 was 846 and 7991, respectively (Table 2).
The sensitivity analysis showed that the two most influential variables in the results obtained for strategies 2 and 3 were the prevalence of MLH1 mutations and the unit cost of a MMR test (Figure 3). For strategy 2, the third most influential variable was the specificity of the hypermethylation, whereas for Strategy 3 was the sensitivity of the hypermethylation. Similar results were obtained when first and second-degree relatives were included in the analysis.

One-way sensitivity analysis for the incremental cost per additional MLH1 mutation detected in probands and in probands and relatives. The three most influential variables, of descending importance, for the incremental cost per additional MLH1 mutation detected for 2 and 3 screening strategy (Panel a and b, respectively). Each horizontal bar shows the range in cost-effectiveness given variations in each parameter value.
Discussion
Lack of BRAF mutations and absence of MLH1 methylation have been proposed as screening tests for the identification of candidate patients for MLH1 germline testing. Here, we show that MLH1 hypermethylation analysis on tumor biopsies, as assessed by MS-MLPA, outperforms BRAF mutation in the selection of these patients in terms of sensitivity and specificity and is more cost-effective.
The association of BRAF mutation with MLH1 hypermethylation and the MSI phenotype resulted in its evaluation as a potential pre-screening tool in the LS diagnostic algorithm 10, 11, 13, 16, 19, 20, 38 (Table 3 and Supplementary Data Table 5). In agreement with previous reports, the sensitivity of the absence of BRAF mutation is very high in identifying MLH1 mutation carriers 16, 18, 21, 39 (Table 3 and Supplementary Data Table 5). A single false-negative was identified adding to the increasing number of LS tumors harboring a BRAF mutation.22 In contrast, its specificity is low. Two factors may account for this observation. First, the low prevalence of BRAF mutations observed in our selected population (11% of MSI tumors and 20% of those lacking MLH1 protein expression). This is in the lower range of reported series 16, 18, 21, 39 but likely to reflect the experience of referral centers.21 Second, the significant number of LS cases and MLH1 germline carriers analyzed allows more accurate estimates.
Sensitivity of methylation of MLH1 promoter was again very high with a single false-negative that also shared a BRAF mutation. The lack of MLH1 promoter hypermethylation showed a sensitivity of 66% for LS depiction. Again, this is in the lower range (57–100%) of reported series 12, 18, 21, 39 (Table 3 and Supplementary Data Table 5) and maybe linked to the low prevalence of hypermethylation observed (42%). This low prevalence may well reflect the demographics of a Cancer Genetics Unit and/or the technique used and the conservative threshold cutoff chosen.
A number of techniques have been proposed to study the hypermethylation in tumor tissues.23, 25, 28 However, we show that MS-MLPA offers a better yield in the routine clinical diagnostic setting,21 since has been a robust methodology, with low variability and good analytical sensitivity when using the highly degraded DNA extracted from FFPE blocks. The definition of clinically meaningful cutoff values is crucial. The arbitrary 20% cutoff value33, 34 has been validated in our series, whereas the 15% threshold21 would have resulted in an increase of false-negative results. Also, we have confirmed that considering only C- and D-regions of MLH1 promoter,36, 40 yields the best performance in the diagnostic setting.18, 21, 39 Quantitative assessment obtained by pyrosequencing is attractive, but its application to FFPE samples is not straightforward.
A false-negative case has been identified. The patient was a carrier of the Spanish pathogenic MLH1 c.1865T>A (L622H) mutation.37 This case shares a BRAF mutation and promoter hypermethylation, the expected scenario for a non-LS tumor.10, 11, 14, 15 Walsh et al22 reported the presence of a BRAF mutation in a member of a LS family that, also showed predisposition to develop colorectal serrated polyps. Interestingly, some evidence suggests that non-LS MSI-H cases may originate from sessile serrated adenoma.41, 42 In our case, no serrated phenotype was observed. The somatic profile of this tumor suggests that hypermethylation is the second inactivating hit. The concomitant existence of BRAF mutations or MLH1 promoter hypermethylation in LS patients has been extensively documented.10, 11, 13, 16, 18, 19, 20, 21, 26, 27, 28
The clinical usefulness of MLH1 hypermethylation analysis relies, in part, on the low prevalence observed. MLH1 hypermethylation analysis does not only outperform BRAF mutation analysis but it is also more cost-effective, in terms of incremental cost per additional MLH1 mutation carrier detected. Our results are in line with those by Perez-Carbonell et al21 that reached similar conclusions using a more simple cost-minimization approach. The advent of Next Generation Sequencing to the diagnostic setting will make germline mutation analysis more affordable. The one-way sensitivity analysis has been used to forecast variations in incremental cost per additional MLH1 mutation carrier. As a token, if a germline analyses would cost 300 per sample the incremental cost per additional mutation carrier detected would be 856 when using MLH1 hypermethtylation as a pre-screening method. This still compares were direct MLH1 germline analysis that associates with an incremental cost of 1620€.
The cost-effectiveness results are also highly sensitive to changes in the prevalence of germline mutation. Our prevalence of 47%, likely reflects the population assessed in referral centers. Also, results are quite sensitive to the operating characteristics of MLH1 methylation detection technique further reinforcing the importance of the technique used. Of note, the recent identification and characterization of MLH1 germline epimutations suggests an additional usefulness of the study of somatic hypermethylation in the diagnostic algorithm of LS eventually depicting those cases candidate for constitutional epigenetic analysis.
The main strengths of our study are: (i) the inclusion of a large series of well-annotated cases with a significant number of LS cases (for a comparison with other studies see Table 3 and Supplementary Data Table 5); (ii) the evaluation of the experience of a Cancer Genetics Unit showing specific prevalence of the alterations; and (iii) the estimation of incremental costs of alternative case-finding methods for detecting MLH1 mutation carriers combined with one-way sensitivity analysis.
In conclusion, somatic hypermethylation of MLH1 is an accurate and cost-effective pre-screening method in the selection of patients that are candidates for MLH1 germline analysis when LS is suspected and MLH1 protein expression is absent. Analysis of MLH1 hypermethylation using MS-MLPA has very few false negative results, making this technique a reasonable option in the diagnostic algorithm of LS. In any case, clinicians must be aware that some LS cases may not be identified. The present study adds significant evidence supporting the introduction of the analysis of somatic hypermethylation of MLH1 as the pre-screening method in the routine diagnostic setting of LS with MLH1 germline mutation.
References
Lynch HT, Krush AJ : Cancer family ‘G’ revisited: 1895-1970. Cancer 1971; 27: 1505–1511.
Mecklin JP, Jarvinen HJ : Tumor spectrum in cancer family syndrome (hereditary nonpolyposis colorectal cancer). Cancer 1991; 68: 1109–1112.
Marra G, Boland CR : Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and historical perspectives. J Natl Cancer Inst 1995; 87: 1114–1125.
Peltomaki P, Vasen HF : Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997; 113: 1146–1158.
Wu Y, Berends MJ, Sijmons RH et al: A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat Genet 2001; 29: 137–138.
Aaltonen LA, Salovaara R, Kristo P et al: Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998; 338: 1481–1487.
Loukola A, Eklin K, Laiho P et al: Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC). Cancer Res 2001; 61: 4545–4549.
Hampel H, Frankel WL, Martin E et al: Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851–1860.
Lindor NM, Burgart LJ, Leontovich O et al: Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol 2002; 20: 1043–1048.
Deng G, Bell I, Crawley S et al: BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res 2004; 10: 191–195.
Domingo E, Laiho P, Ollikainen M et al: BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004; 41: 664–668.
Domingo E, Niessen RC, Oliveira C et al: BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene 2005; 24: 3995–3998.
Lubomierski N, Plotz G, Wormek M et al: BRAF mutations in colorectal carcinoma suggest two entities of microsatellite-unstable tumors. Cancer 2005; 104: 952–961.
McGivern A, Wynter CV, Whitehall VL et al: Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer 2004; 3: 101–107.
Wang L, Cunningham JM, Winters JL et al: BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 2003; 63: 5209–5212.
Bessa X, Balleste B, Andreu M et al: A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol 2008; 6: 206–214.
Domingo E, Espin E, Armengol M et al: Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chromosomes Cancer 2004; 39: 138–142.
Julie C, Tresallet C, Brouquet A et al: Identification in daily practice of patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer): revised Bethesda guidelines-based approach versus molecular screening. Am J Gastroenterol 2008; 103: 2825–2835, quiz 2836.
Lagerstedt Robinson K, Liu T, Vandrovcova J et al: Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst 2007; 99: 291–299.
Loughrey MB, Waring PM, Tan A et al: Incorporation of somatic BRAF mutation testing into an algorithm for the investigation of hereditary non-polyposis colorectal cancer. Fam Cancer 2007; 6: 301–310.
Perez-Carbonell L, Alenda C, Paya A et al: Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn 2010; 12: 498–504.
Walsh MD, Buchanan DD, Walters R et al: Analysis of families with Lynch syndrome complicated by advanced serrated neoplasia: the importance of pathology review and pedigree analysis. Fam Cancer 2009; 8: 313–323.
Bettstetter M, Dechant S, Ruemmele P et al: Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res 2007; 13: 3221–3228.
Cunningham JM, Christensen ER, Tester DJ et al: Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 1998; 58: 3455–3460.
Kuismanen SA, Holmberg MT, Salovaara R, de la Chapelle A, Peltomaki P : Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol 2000; 156: 1773–1779.
Alemayehu A, Sebova K, Fridrichova I : Redundant DNA methylation in colorectal cancers of Lynch-syndrome patients. Genes Chromosomes Cancer 2008; 47: 906–914.
Nagasaka T, Sasamoto H, Notohara K et al: Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol 2004; 22: 4584–4594.
Rahner N, Friedrichs N, Steinke V et al: Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol 2008; 214: 10–16.
Bonis PA, Trikalinos TA, Chung M et al: Hereditary nonpolyposis colorectal cancer: diagnostic strategies and their implications. Evid Rep Technol Assess (Full Rep) 2007; 1–180.
Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009; 11: 35–41.
Motulsky AG : Screening for genetic diseases. N Engl J Med 1997; 336: 1314–1316.
Landi S, Moreno V, Gioia-Patricola L et al: Association of common polymorphisms in inflammatory genes interleukin (IL)6, IL8, tumor necrosis factor alpha, NFKB1, and peroxisome proliferator-activated receptor gamma with colorectal cancer. Cancer Res 2003; 63: 3560–3566.
Berkhout M, Nagtegaal ID, Cornelissen SJ et al: Chromosomal and methylation alterations in sporadic and familial adenomatous polyposis-related duodenal carcinomas. Mod Pathol 2007; 20: 1253–1262.
Jeuken JW, Cornelissen SJ, Vriezen M et al: MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest 2007; 87: 1055–1065.
Azuara D, Rodriguez-Moranta F, de Oca J et al: Novel methylation panel for the early detection of colorectal tumors in stool DNA. Clin Colorectal Cancer 2010; 9: 168–176.
Deng G, Chen A, Hong J, Chae HS, Kim YS : Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res 1999; 59: 2029–2033.
Borras E, Pineda M, Blanco I et al: MLH1 founder mutations with moderate penetrance in Spanish Lynch syndrome families. Cancer Res 2010; 70: 7379–7391.
Rozek LS, Herron CM, Greenson JK et al: Smoking, gender, and ethnicity predict somatic BRAF mutations in colorectal cancer. Cancer Epidemiol Biomarkers Prev 2010; 19: 838–843.
Bouzourene H, Hutter P, Losi L, Martin P, Benhattar J : Selection of patients with germline MLH1 mutated Lynch syndrome by determination of MLH1 methylation and BRAF mutation. Fam Cancer 2010; 9: 167–172.
Hitchins MP, Ward RL : Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet 2009; 46: 793–802.
Dong SM, Lee EJ, Jeon ES, Park CK, Kim KM : Progressive methylation during the serrated neoplasia pathway of the colorectum. Mod Pathol 2005; 18: 170–178.
Kambara T, Simms LA, Whitehall VL et al: BRAF mutation is associated with DNA methyation in serrated polyps and cancers of the colorectum. Gut 2004; 53: 1137–1144.
Acknowledgements
This work was supported by grants from Ministerio de Ciencia e Innovación (SAF 06-06084; 09-07319), Fundació Gastroenterologia Dr Francisco Vilardell (F05-01), Ministerio de Educación y Ciencia Spanish Networks RTICCC (RD06/0020/1050, 1051), Acción en Cáncer (Instituto de Salud Carlos III) and Fundación Científica de la AECC and National Cancer Institute P30 CA46592.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Gausachs, M., Mur, P., Corral, J. et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Hum Genet 20, 762–768 (2012). https://doi.org/10.1038/ejhg.2011.277
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.277
Keywords
This article is cited by
-
Implication of DNA repair genes in Lynch-like syndrome
Familial Cancer (2019)
-
Highly sensitive MLH1 methylation analysis in blood identifies a cancer patient with low-level mosaic MLH1 epimutation
Clinical Epigenetics (2019)
-
Personalized medicine in colorectal cancer diagnosis and treatment: a systematic review of health economic evaluations
Cost Effectiveness and Resource Allocation (2018)
-
BRCA promoter methylation in sporadic versus BRCA germline mutation-related breast cancers
Breast Cancer Research (2017)
-
Precision medicine in heritable cancer: when somatic tumour testing and germline mutations meet
npj Genomic Medicine (2016)
