
Abstract
Although microdeletions of the long arm of chromosome 17 are being reported with increasing frequency, deletions of chromosome band 17q24.2 are rare. Here we report four patients with a microdeletion encompassing chromosome band 17q24.2 with a smallest region of overlap of 713 kb containing five Refseq genes and one miRNA. The patients share the phenotypic characteristics, such as intellectual disability (4/4), speech delay (4/4), truncal obesity (4/4), seizures (2/4), hearing loss (3/4) and a particular facial gestalt. Hallucinations and mood swings were also noted in two patients. The PRKCA gene is a very interesting candidate gene for many of the observed phenotypic features, as this gene plays an important role in many cellular processes. Deletion of this gene might explain the observed truncal obesity and could also account for the hallucinations and mood swings seen in two patients, whereas deletion of a CACNG gene cluster might be responsible for the seizures observed in two patients. In one of the patients, the PRKAR1A gene responsible for Carney Complex and the KCNJ2 gene causal for Andersen syndrome are deleted. This is the first report of a patient with a whole gene deletion of the KCNJ2 gene.
Similar content being viewed by others


Introduction
The human chromosome 17 is characterized by a relative abundance of segmental duplications and interspersed repetitive elements making it prone to genomic rearrangements.1 Hence, various microdeletion and duplication syndromes on chromosome 17 have been reported.2, 3, 4, 5, 6, 7 Deletions of chromosome band 17q24.2 are, however, rare. So far, only nine cases have been reported and five of these were large terminal aberrations ranging from 17q21.3 to qter.8, 9, 10, 11, 12 Common clinical features in these patients included skeletal anomalies, malformations of the palate and/or uvula and of the heart. The other four published cases have been reported with a microdeletion involving chromosome band 17q24.2. The first case, a patient with a 2.3 Mb deletion on 17q24.2-q24.3, was reported by Blyth et al,13 and presented with posterior laryngeal cleft, growth retardation, microcephaly, moderate intellectual disability and numerous freckles and lentigines. In this patient, PRKAR1A (cAMP-dependent protein kinase type I-alpha regulatory subunit), the causal gene for Carney Complex (CNC, OMIM no. 160980), was deleted explaining part of the observed phenotype. The Carney Complex is characterized by pigmented lesions of the skin and mucosae, myxomas, endocrine tumors or endocrine overactivity and schwannomas. Sun et al14 reported three Han Chinese families with congenital generalized hypertrichosis terminalis segregating with a small microdeletion involving chromosome bands 17q24.2-q24.3 for which MAP2K6 (mitogen-activated protein kinase kinase 6) was the best candidate gene. Here we present four previously unreported patients with overlapping microdeletions at chromosome band 17q24.2. Some phenotypic features are shared, making this a new syndromal entity. We delineate the smallest region of overlap (SRO) between all four patients and discuss the genotype–phenotype correlation.
Materials and methods
Clinical reports
Patient 1
The girl was born at term after an uneventful pregnancy as the second child of a healthy unrelated couple. Her birth weight was 3.6 kg and birth length was 52 cm. The head circumference was noted to be large but a documented OFC was not available. She had mild feeding problems with poor weight gain in infancy. She was able to walk at 18 months. She also had speech delay and dyslexia. Her parents mentioned significant anxiety and frequent nightmares and crying during the night. From the age of seven she received special education due to learning difficulties, problems with fine motor development and spatial orientation. At the age of 12 years, a persistent ductus arteriosus was repaired. Cognitive testing at adult age revealed a total IQ of 68, a verbal IQ of 65 and a performance IQ of 76. At the age of 22 years, nocturnal frontal seizures were diagnosed. She presented with visual and auditory hallucinations, uninhibited behavior and anger outbursts. Brain MRI at 23 years showed no structural anomalies apart from a pineal cyst. Ophthalmologic examination showed mild myopia, hearing was normal. She was referred to the genetics center at 23 years of age, because of epilepsy and facial dysmorphism. Genetic analysis revealed a de novo 3 Mb deletion on the long arm of chromosome 17 (46,XX, del(17)(q23.1q23.3)). Clinical examination at the age of 28 years revealed obesity and a muscular build. Her weight was 103 kg (>97th percentile), her length 169.5 cm (50–75th percentile) and her head circumference 63.5 cm (>97th percentile). Facial dysmorphisms consisted of hypertelorism, bushy eyebrows with synophrys, downslanting palpebral fissures, a small upper lip, small but normally positioned ears and irregular tooth implantation. Her neck was broad and short. Her upper limbs showed cubitus valgus and were rather short (span: 157.5 cm). Wrists and ankles were broad. The feet showed bilateral hallux valgus.
Patient 2
Patient 2 is the first child of non-consanguineous parents. Her younger sister is healthy. She was born at 39 weeks gestation with a birth weight of 2.720 kg (10–25th percentile) and length 49.5 cm (50–75th percentile). She was diagnosed with failure to thrive in the first year of life, due to difficult feeding. At age 4 years, her height was 89.3 cm (<3rd percentile) and weight 10.9 kg (<3rd percentile). She was given a presumptive diagnosis of Russell–Silver syndrome and started on growth hormone therapy to which she had a robust response, despite a normal response to growth hormone stimulation testing.
Developmentally, she had significant speech delay in infancy and was subsequently diagnosed with a severe expressive and receptive language disorder. Her speech development was complicated by velopharyngeal insufficiency, for which she underwent a pharyngoplasty at age 5 years. She also had chronic ear infections as a young child and was subsequently diagnosed with a mild-to-moderate conductive hearing loss. Additional medical problems include a history of bilateral grade-2 vesicoureteral reflux, which resolved. At age 10 years, she was discovered to have a prolonged QT interval on an ECG performed in the setting of an episode of syncope. She had a normal cardiac evaluation and genetic testing revealed the p.Asp76Asn mutation in KCNE1 on chromosome 21, which predisposes to Long QT syndrome type 5.15
At age 11 years, her height was 145.4 cm (55th percentile), weight 46.3 kg (84th percentile) and head circumference 52.2 cm (∼50th percentile). She has a triangular face with retrognathia and blepharoptosis. Both ears were small (<−2 SD). Her palate had been noted to be high and arched prior to pharyngoplasty. She had slight clinodactyly of the fifth finger on the left hand. She has very mild scoliosis and a small leg-length inequality, with her right leg being longer than the left. The remainder of her exam was unremarkable.
As an infant, she underwent an initial genetic evaluation, which found a normal 46 XX karyotype as well as normal FISH and methylation studies for Prader–Willi syndrome. She underwent a chromosomal microarray analysis, which revealed a 1.76 Mb de novo deletion spanning 17q24.1–17q24.2.
Patient 3
The girl was delivered at term by an emergency C-section due to decreased fetal movements. Birth weight was 1.720 kg. (<3rd percentile), length and head circumference at birth are not known. Psychomotor development was moderately retarded. Speech was severely delayed with severe impairment in social interaction. From early childhood, she was treated for epilepsy. At 24 years of age, she started having auditory and probably also visual hallucinations. During the same period, she started having panic attacks and mood disturbances. Audiological studies revealed a profound, bilateral predominantly conductive hearing loss. On a CT scan, otosclerosis was observed. She underwent total hip replacement for bilateral avascular necrosis of the femoral head at adult age. Menarche was normal, but she developed secondary amenorrhea. Hypothyroidism became apparent during adolescence and was treated with levothyroxine. Albright hereditary osteodystrophy was initially suspected, but further endocrinological work up performed at the age of 25 years could not confirm this diagnosis (cortisol levels, LH, FSH, parathormone, DHEA, prolactine and IGF1 were normal). Moreover, no mutations could be identified in the GNAS1 gene.
At the age of 28 years, her height was 144.7 cm (<3rd percentile), weight 61.4 kg (10–25th percentile) and head circumference was 53.5 cm (10th percentile). She had short stature, truncal obesity and a seemingly muscular build. The neck was broad and short. Her face was round and flat, with midface hypoplasia, a broad nasal bridge, hypertelorism, deep-set eyes with rather narrow palpebral fissures, full cheeks, rather bulbous nose, short philtrum, small mouth with thin lips and down-turned corners, prognathism and hypodontia with carious teeth. Her hands and feet were small, with tapering fingers, clinodactyly of the fifth fingers, short first toes and syndactyly of the second and third toes (the latter being familial). Ankles and wrists were broad. Her skin was dry, somewhat thickened and ichthyosiform with numerous freckles all over the body, in particular on the face. Multiple striae were noted on her abdomen. Joint movement at the shoulders, wrists and fingers was slightly limited. Elbow extension and flexion were normal, but supination was limited. Karyotype was normal, 46, XX. Many clinical features (muscular build, contractures, short hands, thick skin and hearing loss) were reminiscent of Myhre syndrome (OMIM no. 139210). Array CGH analysis revealed a de novo 2.14 Mb deletion of 17q24.2q24.2.
At the age of 30 years, she was hospitalized because of lethargy. She died at the age of 31 years during an epileptic attack. Autopsy findings showed a large lipomatous tumor in the medullae and cortices of both adrenal glands.
Patient 4
This 2-year old girl presented with symmetrical growth retardation, microcephaly, dysmorphic facial features with a triangular facies, prominent nose, small low-set ears, micrognathia and bilateral 2–3 toe syndactyly. She also had pulmonary stenosis with a dysplastic pulmonary valve, a restrictive persistent ductus arteriosus in association with profound hypotonia and feeding difficulties. She was the second child of non-consanguineous parents, born at 36 weeks weighing 1.8 kg (<3rd percentile). OFC was 33 cm and length 29.6 cm (0.4th–2nd percentiles). She had a stormy neonatal course with inexplicable upper-airway problems and episodes of significant desaturations suggestive of cardiac arrhythmias. Her skin was thin and mottled. Her feeding difficulties were profound with gastroesophageal reflux, necessitating nasogastric tubefeeding and gastrostostomy tube placement. Subsequent growth and development have been significantly delayed. She has delayed dentition and a conductive hearing deficit. She walked independently at the age of 2.5 years, is vocalizing and able to follow simple commands. She has distinctive features with narrow upslanting palpebral fissures, hypertelorism, epicanthus inversus, a prominent nasal bridge, low set ears and micrognathia. The thumbs were broad and in addition she has hypotonia and lax joints. Her facial features have evolved over time, initially being suggestive of Ohdo's syndrome (OMIM no. 249620).
Detailed cardiac assessment in view of the episodes of desaturation showed her to have drug-induced Long QT syndrome. Renal and ophthalmologic investigations were normal.
Array CGH showed a previously unreported de novo 4Mb deletion at 17q24.2q24.3.
Cytogenetic analysis
Conventional karyotyping of G-banded metaphase chromosomes was performed on short-term cultured lymphocytes using standard procedures. No visible structural anomalies were detected in any of the probands.
Array CGH analysis
All microdeletions were detected using genomic microarrays. To further fine map the deletions, array CGH was performed on patient 1, 3 and 4 using a custom 8 × 60K oligonucleotide array (Agilent Technologies, Santa Clara, CA, USA). These arrays covered the following region chr17:61158508–69685732 (hg19) with a total of 52474 probes. Blyth et al13 reported the first case with a 17q24.2 microdeletion investigated by genomic microarrays. As there is some phenotypic overlap between the four patients reported here and this patient, the patient from Blyth et al13 was also included to refine the breakpoints. The assays were performed as previously described.16 Patient 2 had a custom 105K chromosomal microarray performed at the Medical Genetics Laboratory of Baylor College of Medicine.
Sequencing analysis
All coding sequences of the protein-coding genes (PRKCA, CACNG5, CACNG4, CACNG1 & HELZ) located in the SRO were amplified and subsequently sequenced using the ABI 3730XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Primer sequences and PCR conditions are available upon request. The first exons of PRKCA and CACNG4 could not be amplified due to the high GC content.
Results
In all patients a small interstitial deletion involving chromosomal band 17q24.2 was identified with molecular karyotyping. Array CGH results of all patients are summarized in Table 1 (GRCh37 February 2009 assembly). The SRO is only 713 kb in size and contains 1 microRNA and 5 RefSeq genes; PRKCA, HELZ and a cluster of three CACNG genes encoding the gamma subunit of a voltage-dependent calcium channel (Figure 1). Patient 2's parents had FISH testing performed for the involved region yielding a normal result meaning that her deletion originated de novo. For the other patients, array CGH analysis of the parents yielded normal results, confirming the de novo status of the deletions.

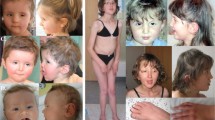
(a) Overview of the clinical features of all four patients. (b) Overview of the overlapping 17q24.2 microdeletions in patients 1–4 and the deletion in the patient reported by Blyth et al. Breakpoint locations were uploaded into the UCSC Genome Browser (February 2009 assembly). The genes causal for Carney Complex (PRKAR1A) and Andersen Syndrome (KCNJ2) are highlighted. ID: intellectual disability.
As many clinical features of patient 3 were reminiscent of Myhre syndrome (OMIM no. 139210), sequencing analysis of the deleted genes in the SRO was performed on patient 3, as well as on two other unrelated patients suspected to have Myhre syndrome. No mutations could however be detected in the coding region of these genes in any of these three patients. In addition, array CGH analysis revealed normal results in the two unrelated patients with Myhre syndrome.
Discussion
Chromosome 17 harbors several known microdeletion and microduplication syndromes. Microdeletions of chromosome band 17q24.2 are however rare. Here we present four patients with an overlapping deletion on chromosome band 17q24.2 and a strong phenotypic resemblance. The clinical features seen in all our four patients consist of developmental delay, speech delay, feeding problems in infancy, truncal obesity and a similar facial gestalt. Of the nine cases previously reported with 17q24.2 deletions, five had large cytogenetically detectable deletions ranging from 17q21.3 to 17qter.8, 9, 10, 11, 12 Common clinical features in these patients included skeletal anomalies, malformations of the palate and/or uvula and cardiac malformations. Most of them were infants who died within the first few months, so the childhood phenotype and developmental assessments are not available.
The breakpoints in the four patients presented in this paper do not cluster together nor are they flanked by low-copy repeats or homologs LINEs (long interspersed repetitive elements) or SINEs (short interspersed repetitive elements), suggesting that these rearrangements were not mediated by non-allelic homologous recombination. None of these deletions have been reported in the Toronto Database of Genomic Variants (http://projects.tcag.ca/variation) nor in the Redon AffyStudy.17
Some of the clinical features observed in our patients are in common with patients with Myhre syndrome (OMIM no. 139210). This is a rare syndrome characterized by blepharophimosis, a muscular build, deafness, brachydactyly, stiff skin and limited joint mobility. Only 16 patients have been reported with this syndrome to date.18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 As patients with larger overlapping deletions do not have clear characteristics of Myhre syndrome and recessive inheritance for Myhre syndrome cannot be excluded, sequencing analysis of the coding genes residing in the SRO was performed on patient 3, who had the most resemblance with Myhre syndrome, as well as on two other unrelated patients with suspicion of Myhre syndrome. No mutations were however identified.
As there is an interesting clinical overlap between the presented patients, the following conclusions regarding genotype–phenotype correlations can be made.
Facial phenotype
The patients reported here have a striking facial resemblance. This consists of small ears (4/4), hypertelorism (4/4), a bulbous nose (3/4), a thin (upper) lip (4/4), a flat round face (3/4), sparse eyebrows (3/4) and pro- or micrognathism (3/4) (Figure 1).
Hearing loss
Patients 2, 3 and 4 all present with conductive hearing loss. For all three patients, repeated ear infections were noted at younger age, which may give an explanation for the hearing loss.
Staturoponderal development
Short stature was noted in all patients except for patient 1. Patient 2 was given growth hormone replacement therapy and showed a robust response.
The typical body shape with truncal obesity is a common feature seen in all patients. One interesting gene in the SRO is the protein kinase C alpha gene (PRKCA). PRKCA encodes the serine- and threonine-specific protein kinase C alpha that has an important role in many different cellular processes. In 2009, a genome-wide linkage analysis of BMI (body mass index) appointed PRKCA as a pleiotropic locus to be associated with BMI and asthma.29 Deletion of PRKCA might contribute to the observed truncal obesity. However, more studies are needed to further investigate this possible role.
Endocrinologic abnormalities
One of the deleted genes in patient 4 is the PRKAR1A gene. This gene is causal for the autosomal dominant Carney Complex (CNC, OMIM no. 160980).30, 31, 32 This complex is characterized by pigmented lesions of the skin and mucosae, myxomas, endocrine tumors or overactivity and schwannomas. The patient of Blyth et al13 was reported with Carney Complex Plus syndrome, indicating that not all her phenotypic characteristics could be explained by Carney Complex. She presented with growth retardation, posterior laryngeal cleft, numerous freckles and lentigines, microcephaly and moderate intellectual disability.
The typical cutaneous lesions of Carney complex are not present in patient 4 and an echocardiography showed no myxomas thus far. No further endocrinologic data are available for this patient. Although detectable at an early age, most endocrine tumors in Carney complex become clinically significant during the second decade of life.33
Although the endocrinological work up at the age of 25 years was normal in patient 3, the presence of a large tumor of the adrenal glands (detected on autopsy) and numerous freckles all over the body, in particular on the face, are signs possibly associated with Carney complex, but are not diagnostic for the disease.33 The PRKAR1A gene is not deleted in patient 3, it is however possible that regulatory elements in this deletion may have an influence on PRKAR1A expression and as a consequence on the phenotype. As this patient is deceased, evaluation for other signs of the Carney complex and for PRKAR1A expression is not possible anymore.
Skin and teeth
The suspicion of Myhre syndrome was most striking for patient 3, as she also had a thick and ichthyosiform skin, a feature commonly observed in patients with Myhre syndrome. Patient 3 also presented with numerous freckles all over the body, in particular on the face. Patient 4 has a mottled/translucent skin. The freckles and lentigines typical for Carney Complex were not yet seen in patient 4.
Delayed dentition was noted for patients 3 and 4, and was also described for the patient reported by Blyth et al.13 The deleted region shared by these three patients is 649 kb in size, and contains eight RefSeq genes (BPTF (disrupted), KPNA2, C17orf58, loc100499466, loc440641, AMZ2, ARSG and SLC16A6). None of these genes have been directly linked to teeth development. Kpna2 (karyopherin alpha 2) however, is required for the import of dmp1 (dentin matrix protein 1) into the nucleus.34 It has been suggested that DMP-1 is essential for dentinogenesis during postnatal development.35 Blyth et al13 suggested that deletion of KPNA2 accounted for the phenotypic characteristics of their patient (short stature, microcephaly, syndactyly, clinodactyly and intellectual disability), which could not be explained by Carney Complex.13, 36 In patient 4, the contribution of KCNJ2 (Inward rectifier potassium channel 2, subfamily J), for which heterozygous mutations are reported to be responsible for Long QT syndrome type 7 also known as Andersen syndrome (OMIM no. 170390), cannot be excluded. This syndrome includes dental defects, such as persistent primary teeth, delayed eruption of secondary teeth, oligodontia, elongated roots with open apices and enamel hypoplasia.
Cardiac malformations
Patient 1 had a persistent ductus arteriosus that was repaired at the age of 12 years. Patient 2 has a prolonged QT interval caused by a mutation in the KCNE1 gene on chromosome 21, and patient 4 presents with a drug-induced prolonged QT interval. As mentioned earlier, the deletion of patient 4 includes KCNJ2 of which heterozygous mutations are causal for Andersen syndrome.37 This syndrome is characterized by periodic paralysis, ventricular arrhythmias and prolonged QT intervals, dental anomalies, small hands and feet, low-set ears, hypertelorism, micrognathia, fifth-digit clinodactyly and syndactyly. Also, joint laxity has been reported. Most of the clinical features seen in patient 4 (and not in the other patients) can be explained by this syndrome, including the arrhythmias, the drug-induced prolonged QT, the low-set ears and the hypotonia/lax joints. This patient is the first case reported with Andersen syndrome to have a whole gene deletion.
Psychomotor development
In 2006, de Quervain and Papassotiropoulos38 found a significant association (P=0.00008) between short-term episodic memory performance and genetic variations in a seven-gene cluster containing PRKCA. Jablensky et al39 also showed a contribution of PRKCA to dysfunctional memory performance in schizophrenia with pervasive cognitive deficits. This indicates that PRKCA is important for human memory function, suggesting that the deletion of PRKCA might be involved in the mild-to-moderate intellectual disability seen in these four patients.
Of note, in patients 1 and 3, abnormal behavior was noted. Both patients presented with visual and auditory hallucinations together with mood disturbances. The deletions reported in these two patients have an overlap of approximately 982 kb. In this region, PRKCA, CACNG4, CACNG1 and CACNG5 have been implicated directly or indirectly in the pathogenesis of psychiatric disorders. Recently, SNPs in PRKCA have been described to be associated with schizophrenia.40 Next to PRKCA, lies a cluster of three CACNG genes encoding the gamma subunit of a voltage-dependent calcium channel. This type of calcium channel exists of four subunits (alpha, beta, gamma and alpha2–delta). Genes encoding the other subunits have already been associated with psychiatric disorders,41, 42 and Curtis et al43 recently suggested that voltage-dependent calcium channel genes could have an important role in the susceptibility to bipolar disorder and/or schizophrenia, in particular CACNG5. Both PRKCA and this cluster are involved in the CREB signaling pathway in neurons. Hence, deletion of (one of) these genes might be responsible for the mood swings and hallucinations seen in patients 1 and 3. It is not clear whether the deletion of a single gene or the deletion of all these genes contributes to the phenotype. Patient 2 and 4 do not have these features despite the overlapping deletion, indicating that the deletion is probably a susceptibility factor rather than a direct cause. Patient 4 is still young, and as such, it cannot be ruled out that she will develop these features later in time.
Seizures were noted in patients 1 and 3. In patient 1, these seizures were nocturnal. Genes encoding the alpha, beta and alpha2–delta subunit of voltage-gated calcium channels have already been associated with epilepsy.44, 45 For both patients, it is possible that the deletion of the CACNG cluster is also a susceptibility factor for their seizures.
Conclusion
In conclusion, we report four patients with overlapping deletions involving chromosome band 17q24.2, with a SRO of 713 kb. The patients share some interesting clinical features, such as intellectual disability with a pronounced speech delay, truncal obesity and a similar facial gestalt making this a new syndromal entity. Protein kinase C represents a family of closely related enzymes that phosphorylates residues of various intracellular proteins, and is thus involved in a wide range of cellular functions, such as ion transport, release of hormones or neurotransmitters, downregulation of receptors or expression of various proteins.46 Hence, deletion of the PRKCA gene might be responsible for many of the observed features, such as the intellectual disability, the truncal obesity, the mood swings and hallucinations. The latter could however also been caused by deletion of the CACNG cluster, which could also account for the seizures observed in two patients. Delayed dentition is seen in two patients and in the patient reported by Blyth et al,13 suggesting that deletion of KPNA2 could account for it. Finally, patient 4 is the first patient to be reported with a whole gene deletion leading to Andersen syndrome (KCNJ2). To further strengthen these genotype–phenotype correlations more patients with overlapping deletions need to be investigated.
References
Zody MC, Garber M, Adams DJ et al: DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature 2006; 440: 1045–1049.
Koolen DA, Vissers LE, Pfundt R et al: A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 2006; 38: 999–1001.
Lupski JR, de Oca-Luna RM, Slaugenhaupt S et al: DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991; 66: 219–232.
Chance PF, Alderson MK, Leppig KA et al: DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993; 72: 143–151.
Smith AC, McGavran L, Robinson J et al: Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet 1986; 24: 393–414.
Bi W, Sapir T, Shchelochkov OA et al: Increased LIS1 expression affects human and mouse brain development. Nat Genet 2009; 41: 168–177.
Riva P, Corrado L, Natacci F et al: NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet 2000; 66: 100–109.
Bridge J, Sanger W, Mosher G et al: Partial duplication of distal 17q. Am J Med Genet 1985; 22: 229–235.
Dallapiccola B, Mingarelli R, Digilio C, Obregon MG, Giannotti A : Interstitial deletion del(17) (q21.3q23 or 24.2) syndrome. Clin Genet 1993; 43: 54–55.
Thomas JA, Manchester DK, Prescott KE, Milner R, McGavran L, Cohen Jr MM : Hunter-McAlpine craniosynostosis phenotype associated with skeletal anomalies and interstitial deletion of chromosome 17q. Am J Med Genet 1996; 62: 372–375.
Olney PN, Kean LS, Graham D, Elsas LJ, May KM : Campomelic syndrome and deletion of SOX9. Am J Med Genet 1999; 84: 20–24.
Levin ML, Shaffer LG, Lewis R, Gresik MV, Lupski JR : Unique de novo interstitial deletion of chromosome 17, del(17) (q23.2q24.3) in a female newborn with multiple congenital anomalies. Am J Med Genet 1995; 55: 30–32.
Blyth M, Huang S, Maloney V, Crolla JA, Karen Temple I : A 2.3Mb deletion of 17q24.2-q24.3 associated with ‘Carney Complex plus’. Eur J Med Genet 2008; 51: 672–678.
Sun M, Li N, Dong W et al: Copy-number mutations on chromosome 17q24.2-q24.3 in congenital generalized hypertrichosis terminalis with or without gingival hyperplasia. Am J Hum Genet 2009; 84: 807–813.
Schulze-Bahr E, Haverkamp W, Funke H : The long-QT syndrome. N Engl J Med 1995; 333: 1783–1784.
Buysse K, Delle Chiaie B, Van Coster R et al: Challenges for CNV interpretation in clinical molecular karyotyping: lessons learned from a 1001 sample experience. Eur J Med Genet 2009; 52: 398–403.
Redon R, Ishikawa S, Fitch KR et al: Global variation in copy number in the human genome. Nature 2006; 444: 444–454.
van Steensel MA, Vreeburg M, Steijlen PM, de Die-Smulders C : Myhre syndrome in a female with previously undescribed symptoms: further delineation of the phenotype. Am J Med Genet A 2005; 139A: 127–130.
Becerra-Solano LE, Diaz-Rodriguez M, Nastasi-Catanese JA et al: The fifth female patient with Myhre syndrome: further delineation. Clin Dysmorphol 2008; 17: 113–117.
Burglen L, Heron D, Moerman A et al: Myhre syndrome: new reports, review, and differential diagnosis. J Med Genet 2003; 40: 546–551.
Davalos NO, Garcia-Ortiz JE, Garcia-Cruz D, Feria-Velasco A, Sanchez-Corona J : Myhre syndrome: first female case. Clin Dysmorphol 2003; 12: 119–121.
Garcia-Cruz D, Figuera LE, Feria-Velazco A et al: The Myhre syndrome: report of two cases. Clin Genet 1993; 44: 203–207.
Lopez-Cardona MG, Garcia-Cruz D, Garcia-Ortiz JE et al: Second female case of Myhre syndrome. Clin Dysmorphol 2004; 13: 91–94.
Titomanlio L, Marzano MG, Rossi E et al: Case of Myhre syndrome with autism and peculiar skin histological findings. Am J Med Genet 2001; 103: 163–165.
Whiteford ML, Doig WB, Raine PA, Hollman AS, Tolmie JL : A new case of Myhre syndrome. Clin Dysmorphol 2001; 10: 135–140.
Myhre SA, Ruvalcaba RH, Graham CB : A new growth deficiency syndrome. Clin Genet 1981; 20: 1–5.
Soljak MA, Aftimos S, Gluckman PD : A new syndrome of short stature, joint limitation and muscle hypertrophy. Clin Genet 1983; 23: 441–446.
Rulli I, Ferrero GB, Belligni E, Delmonaco AG, Defilippi C, Silengo M : Myhre's syndrome in a girl with normal intelligence. Am J Med Genet A 2005; 134A: 100–102.
Murphy A, Tantisira KG, Soto-Quiros ME et al: PRKCA: a positional candidate gene for body mass index and asthma. Am J Hum Genet 2009; 85: 87–96.
Kirschner LS, Carney JA, Pack SD et al: Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000; 26: 89–92.
Horvath A, Bossis I, Giatzakis C et al: Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 2008; 14: 388–395.
Horvath A, Bertherat J, Groussin L et al: Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 2010; 31: 369–379.
Rothenbuhler A, Stratakis CA : Clinical and molecular genetics of Carney complex. Best Pract Res Cl En 2010; 24: 389–399.
Narayanan K, Ramachandran A, Hao J et al: Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. J Biol Chem 2003; 278: 17500–17508.
Ye L, MacDougall M, Zhang S et al: Deletion of dentin matrix protein-1 leads to a partial failure of maturation of predentin into dentin, hypomineralization, and expanded cavities of pulp and root canal during postnatal tooth development. J Biol Chem 2004; 279: 19141–19148.
Tseng SF, Chang CY, Wu KJ, Teng SC : Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J Biol Chem 2005; 280: 39594–39600.
Plaster NM, Tawil R, Tristani-Firouzi M et al: Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001; 105: 511–519.
de Quervain DJ, Papassotiropoulos A : Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proc Natl Acad Sci USA 2006; 103: 4270–4274.
Jablensky A, Morar B, Wiltshire S et al: Polymorphisms associated with normal memory variation also affect memory impairment in schizophrenia. Genes Brain Behav 2011; 10: 410–417.
Carroll LS, Williams NM, Moskvina V et al: Evidence for rare and common genetic risk variants for schizophrenia at protein kinase C, alpha. Mol Psychiatry 2010; 15: 1101–1111.
Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT : CACNA1H mutations in autism spectrum disorders. J Biol Chem 2006; 281: 22085–22091.
Ferreira MA, O’Donovan MC, Meng YA et al: Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 2008; 40: 1056–1058.
Curtis D, Vine AE, McQuillin A et al: Case-case genome-wide association analysis shows markers differentially associated with schizophrenia and bipolar disorder and implicates calcium channel genes. Psychiatr Genet 2011; 21: 1–4.
Liang J, Zhang Y, Chen Y et al: Common polymorphisms in the CACNA1H gene associated with childhood absence epilepsy in Chinese Han population. Ann Hum Genet 2007; 71: 325–335.
Stam AH, Luijckx GJ, Poll-The BT et al: Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry 2009; 80: 1125–1129.
Newton AC : Protein kinase C: structure, function, and regulation. J Biol Chem 1995; 270: 28495–28498.
Acknowledgements
We are grateful to all patients, their families and the clinicians involved for their cooperation. We would like to thank Lies Vantomme, Shalina Baute and Elke Van Oudenhove for their expert technical assistance. Sarah Vergult is supported by a Ph.D. fellowship of the Research Foundation – Flanders (FWO). This work was supported by grant SBO60848 from the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT), grant G.0279.07N of the FWO and a Methusalem grant of the Flemish Government. This article presents research results of the Belgian program of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming (IUAP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Vergult, S., Dauber, A., Chiaie, B. et al. 17q24.2 microdeletions: a new syndromal entity with intellectual disability, truncal obesity, mood swings and hallucinations. Eur J Hum Genet 20, 534–539 (2012). https://doi.org/10.1038/ejhg.2011.239
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.239
Keywords
This article is cited by
-
Transcriptomics unravels molecular players shaping dorsal lip hypertrophy in the vacuum cleaner cichlid, Gnathochromis permaxillaris
BMC Genomics (2021)
-
Family-Based Whole Genome Sequencing Identified Novel Variants in ABCA5 Gene in a Patient with Idiopathic Ventricular Tachycardia
Pediatric Cardiology (2020)
-
Chromosomal microarray analysis in the genetic evaluation of 279 patients with syndromic obesity
Molecular Cytogenetics (2018)
-
Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability
European Journal of Human Genetics (2015)
-
Investigation of selected genomic deletions and duplications in a cohort of 338 patients presenting with syndromic obesity by multiplex ligation-dependent probe amplification using synthetic probes
Molecular Cytogenetics (2014)
