
Abstract
The correct interpretation of copy number gains in patients with developmental delay and multiple congenital anomalies is hampered by the large number of copy number variations (CNVs) encountered in healthy individuals. The variable phenotype associated with copy number gains makes interpretation even more difficult. Literature shows that inheritence, size and presence in healthy individuals are commonly used to decide whether a certain copy number gain is pathogenic, but no general consensus has been established. We aimed to develop guidelines for interpreting gains detected by array analysis using array CGH data of 300 patients analysed with the 105K Agilent oligo array in a diagnostic setting. We evaluated the guidelines in a second, independent, cohort of 300 patients. In the first 300 patients 797 gains of four or more adjacent oligonucleotides were observed. Of these, 45.4% were de novo and 54.6% were familial. In total, 94.8% of all de novo gains and 87.1% of all familial gains were concluded to be benign CNVs. Clinically relevant gains ranged from 288 to 7912 kb in size, and were significantly larger than benign gains and gains of unknown clinical relevance (P<0.001). Our study showed that a threshold of 200 kb is acceptable in a clinical setting, whereas heritability does not exclude a pathogenic nature of a gain. Evaluation of the guidelines in the second cohort of 300 patients revealed that the interpretation guidelines were clear, easy to follow and efficient.
Similar content being viewed by others

INTRODUCTION
High-resolution genome-wide array analysis enables the detection of submicroscopic copy number variations (CNVs), as small as only a few kilobases. Using array, an extra 15% causally related chromosomal abnormalities are detected over routine microscopic and MLPA subtelomeric screening in patients with developmental delay (DD) and/or multiple congenital anomalies (MCAs).1 However, understanding the clinical relevance of CNVs is lagging behind the rapid increase in resolution of this genome-wide screening technique. The presence of large numbers of CNVs with no major phenotypic effect impede the interpretation of array results in DD/MCA patients.2, 3 Interpreting copy number gains appears even more complicated than interpreting losses. It is generally assumed that microduplications tend to have a milder and more variable phenotype.4 Moreover, the gain-of-function effect of genes is less often known than their loss-of-function effect.
The rule that de novo chromosomal imbalances are most likely to be clinically significant, whereas familial CNVs are not, does not always hold true. Several studies have shown the clinical relevance of inherited CNVs and therefore the de novo origin of a CNV is not a good indicator of its clinical relevance.5, 6, 7 A more reliable way of determing the clinical relevance of a CNV is to compare it with CNVs gathered in large databases with data of healthy controls. The Database of Genomic Variants (http://projects.tcag.ca/variation) is a well-known database. Several laboratories also have available an in-house or national reference database. The ‘Low Lands consortium’ reference database was developed as a joint venture of five Dutch laboratories, using the same Agilent 105K oligo array. At the starting point of this study, the database contained CNVs from more than 300 healthy parents of probands, but it grew rapidly during the course of the study to more than 700. Despite these helpful databases, the clinical significance of many CNVs remains unknown.
Hitherto, four published studies present a structured interpretation of CNVs in patients with DD and/or MCA.8, 9, 10, 11 These studies included both copy number losses and gains. Koolen et al8 stated in their interpretation workflow that if a CNV is familial, it is likely not to be clinically relevant. However, as mentioned above, this approach is debatable. Gijsbers et al9 used a slightly different approach. Syndromic CNVs were considered clinically relevant, regardless of whether they were de novo or not. However, in the remaining group of potentially relevant CNVs, inherited CNVs were considered as not likely to be clinically relevant. Buysse et al10 used a comparable approach. In their first step, CNVs which were related to known microduplication and microdeletion syndromes, or were known DD/MCA loci, were considered causal. In the second step, they concluded all common CNVs were probably not relevant. In their last step, all de novo gains were considered causal, whereas inherited gains were considered of unknown clinical significance. Hence, in their last step, they concluded the effect of the remaining gains based entirely on the origin of the CNVs. In the fourth study, Bruno et al11 applied a comparable way of analysing CNVs, based on the guidelines described by Lee et al.12 Bruno et al11 mentioned that they did not exclusively apply a de novo origin of a CNV as a criterion for clinical relevance. This was not further explained, so it is difficult to see how they interpreted individual cases. So far, the only study focusing exclusively on copy number gains was published by Stankiewicz et al,13 but their paper described only a few examples of well-analysed gains.
The aim of our study was to develop practical guidelines for the clinical interpretation of copy number gains. We evaluated all gains in a cohort of 300 DD/MCA patients using an interpretation scheme and correlated their clinical relevance to the origin and size of the gains. We evaluated different size thresholds for the detection of gains in routine diagnostics. On the basis of our results, we drew up guidelines and evaluated them in a second, independent, cohort of 300 DD/MCA patients.
PATIENTS AND METHODS
Patients, parents and controls
The first 300 patients analysed by high-resolution array CGH in our department were included. Patients were referred because of the presence of DD, behavioural problems and/or congenital anomalies. Their parents were investigated by array CGH, whenever available. None of the investigated parents had a clinical phenotype resembling that of their offspring.
A second cohort of 300 independent DD/MCA patients, referred during the first 4 months of 2009, was used to evaluate our guidelines.
The data of healthy individuals in the Low Lands consortium reference database (Nexus 4.0; Bio Discovery, Inc., El Segundo, CA, USA) were used as a control group. At the beginning of the study, this database contained information on over 300 healthy parents. During the second phase, over 700 controls were included.
Array comparative genomic hybridisation
Array CGH was performed using the 105K oligo array Oxford Design from Agilent (custom design ID: 019015; Agilent Technologies Inc., Santa Clara, CA, USA). A mixture of 40 healthy male or female DNA samples was used as a reference (sex-matched). Procedures were performed according to the manufacturer's protocol. Data were extracted using Feature Extraction V.9.1 software (Agilent Technologies Inc.). An array was classified as successful if the Derivative of Log Ratio Standard deviation was below 0.20 and the raw array CGH data of the first 300 successful arrays were analysed for the presence of gains using DNA analytics (Agilent Technologies Inc.), using the ADM-2 aberration algorithm. Alterations were concluded to be a significant gain if at least four adjacent probes had an average log ratio of at least 0.4. Gains larger than 10 Mb were not considered as microduplications and were excluded from further analysis. Gains were analysed according to hg18 (NCBI Build 36.1; University of California-Santa Cruz Human Genome Browser, http://genome.ucsc.edu/).
Interpretation of gains
An interpretation scheme to determine the clinical relevance of the detected gains was developed. The scheme is partly based on previously published studies,8, 9, 10, 11 but did not include origin or size as possible exclusion criterium, as these were subject of our study in the first cohort. We assessed the gains of this cohort using the following steps:
Step 1. Comparison with the Low Lands consortium reference database. Some of the healthy parents from the patients included in this study were already part of this anonymous control data set, hence we decided to set the minimum number of gains that had to be present in the database before concluding a gain was benign, at four instead of three (1%), which is routinely used. We concluded that all the gains present in this database ≥4 times, or three times together with ≥5 times their reciprocal loss, were benign CNVs.
Step 2. Comparison with the Database of Genomic Variants. All gains present in this independent database ≥3 times, or two times together with ≥5 times their reciprocal loss, were considered to be benign CNVs.
Step 3. Collection of detailed clinical data and comparison with known microduplication syndromes. If a gain was involved in a known microduplication syndrome (see syndrome list of Decipher: http://decipher.sanger.ac.uk/) and the clinical features of the patient were in accordance with this syndrome, we considered the gain was clinically relevant.
Step 4. For the remaining gains, we searched Genatlas (http://genatlas.medecine.univ-paris5.fr) and the UCSC browser (http://genome.ucsc.edu) for the presence and function of genes located in the gains. If no genes were present in the gain, or only genes with known function irrelevant to the clinical phenotype of the patient, we concluded the gain was a benign CNV.
Step 5. For the remaining gains (ie, those with possibly relevant genes or genes with unknown function), we searched for cases with comparable microduplications using the PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), Embase (http://www.embase.com/), Decipher (http://decipher.sanger.ac.uk) and ECARUCA (http://www.ecaruca.net). If a duplication in the same area or wider surrounding area, with a partly or comparable clinical phenotype, was found, we concluded the gain was clinically relevant. If no overlapping duplications were found, or duplications with a different phenotype, we concluded the gain as a CNV of unknown clinical relevance.
Thus, the possible outcomes of our interpretation scheme are: a clinically relevant CNV, a CNV of unknown clinical relevance or a benign CNV.
Evaluation of the guidelines
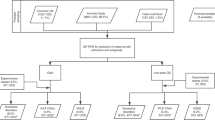
We designed a flow diagram (Figure 1) for gains with a threshold of 200 kb, based on our results in the first cohort of 300 patients. We used the second cohort of 300 DD/MCA patients for the evaluation.

Flow diagram for interpreting gains based on the results of this study. *Confirm location of duplication with FISH.
Statistical analysis
Statistical calculations were performed using the Statistical Package for the Social Sciences version 17.0 for Windows (SPSS Inc., Chicago, IL, USA) and the following tests were performed whenever appropriate: Binomial test, Mann–Whitney U-test, Pearson χ2-test and Student's t-test. A P-value <0.05 was considered significant.
RESULTS
Interpretation of gains in the first 300 patients
A total number of 805 gains of at least four adjacent oligonucleotides were detected in the first cohort of 300 patients. Three of these gains were 91, 64 and 21 Mb in size and were excluded from further analysis. Another four gains in two patients with a 47,XYY karyotype were excluded because they comprised the pseudoautosomal regions of the Y chromosome. One other gain of 5.5 Mb was excluded because it was detected in a patient with an unbalanced translocation der(12)t(9;12)(q34.13;p13.32), in which the accompanying deletion explained the phenotype. We finally included a total number of 797 gains (Supplementary Table 1), detected in 287 different patients. Only 13 patients did not have any gains.
The intepretation results are summarised in Table 1. In short, 546 out of 797 gains (68.5%) were benign CNVs because of their presence in the reference database. Of the remaining 251 gains, 151 were benign CNVs (60.2%) because of their presence in the Database of Genomic Variants. A further eight gains were associated with known microduplication syndromes (1q21, 15q11q13, 16p11.2, 22q11.2 (four times) and Xq28) (http://decipher.sanger.ac.uk). On the basis of the information from the genome browsers and the literature, we considered 7 additional gains to be clinically relevant and 29 gains to be benign. One maternally inherited 253 kb gain of exons 45–50 of the DMD gene (Xp21.1) was seen in a boy and confirmed by MLPA. A tandem intragenic duplication of these exons is known to result in a truncated protein. However, the boy had mild mental retardation, but no clinical features of Duchenne muscular dystrophy and normal creatin kinase levels. FISH analysis showed that the duplication was inserted in Xq27 and did not disrupt the DMD gene. As the maternally inherited insertion might have a positional effect at Xq27, this was considered a CNV of unknown clinical relevance.
We finally concluded that 726 (91.1%) gains were benign, 15 (1.9%) were clinically relevant and the remaining 56 (7.0%) were of unknown clinical relevance. Supplementary Table 2a gives an overview of the location, size and origin of the 15 clinically relevant gains and the phenotypes of the patients.
Assessing the origin of gains in the first cohort
The origin could be established in 508 out of 797 gains (63.7%); 230/508 (45%) were de novo and 278/508 (55%) were familial (Table 2). There were significantly more familial gains than de novo gains (binomial test, P=0.037). The origin was known for 14 of the 15 clinically relevant gains (Supplementary Table 2a). More clinically relevant gains were familial (10/14; 71%) than de novo (4/14; 29%). In contrast, benign gains were identified only slightly more often as familial (242/460; 53%) than de novo (218/460; 47%). Heritability was not significantly different between clinically relevant and benign gains (Pearson χ2-test, P=0.20).
Determination of a practical size threshold in the first cohort
The average size of clinically relevant gains was 2283 kb (range 288–7912 kb) (Table 3). This was significantly different from the size of benign gains and those of unknown relevance (Mann–Whitney U-test, P<0.001). The wide size range of benign gains is caused by a duplication of 7.94 Mb in 9p13p11. The pericentromeric 9q region is known to be highly variable without having clinical consequences.14
In Table 4, the effects of thresholds of 0 (but with at least four adjacent oligonucleotides), 100, 200, 300, 400 and 500 kb are shown. With a threshold of 200 kb, none of the relevant gains, 18 gains of unknown clinical relevance and 436 benign gains would have remained undetected (100% sensitivity for the relevant gains). At this threshold, 84.5% (290/343) of all the detected gains are benign CNVs (specificity 15.5%). Increasing the threshold to 300, 400 or 500 kb hardly affects the specificity but it does decrease the sensitivity. On the other hand, a lower threshold reduces the specificity without increasing the sensitivity. For example, at a threshold of 100 kb, 617 out of 682 gains (90.5%) are benign vs 290 out of 343 gains (84.5%) at 200 kb (t-test, P=0.005) (Table 4).
Evaluation of the interpretation scheme
After assessing all detected gains in the first 300 patients, we designed a flow diagram of our interpretation scheme (Figure 1). A threshold of 200 kb was added because of its favourable sensitivity and specificity as determined above. To increase the reliability of the decision based on the control data sets, we used a 1% threshold for our rapidly expanding reference database, at that moment containing over 700 controls, and at least three different studies (BAC CNVs excluded) for the Database of Genomic Variants. This flow diagram was evaluated using a second cohort of 300 DD/MCA patients.
In the second cohort we detected 598 gains over 200 kb in size. Four gains were larger than 10 Mb and therefore excluded. The interpretation results of the remaining 594 gains are summarised in Table 1. In total, 506 (85.2%) of the gains were considered benign, 72 (12.1%) were of unknown clinical relevance and 16 (2.7%) were clinically relevant (Supplementary Table 2b). The inheritence pattern could be established for 12 relevant gains: six were familial (including one X-linked) and six were de novo (including one X-chromosomal). The results in the second cohort are comparable to the interpretation results for the 343 gains above 200 kb detected in the initial study group, with 290 (84.5%) classified as benign, 38 (11.1%) as unknown and 15 (4.4%) as clinically relevant CNVs (Tables 1 and 4).
DISCUSSION
In this study we focused on interpreting copy number gains detected by genome-wide array analysis in patients with DD/MCA. Combining literature and our laboratory findings, we developed an interpretation scheme for copy number gains. We did not exclude patients in whom a clinically relevant loss was detected, as we considered gains as independent events that should be interpreted independently. After evaluating all the gains, three patients with a clinically relevant gain also had accompanying deletions that may have contributed to their phenotypes (patients 11, 16 and 27; Supplementary Table 2). Further, two patients had proven mutations in other disease-causing genes (patients 12 and 21). We believe, however, that the duplications may have contributed to their phenotypes, as illustrated by patient 12, who had a molecularly confirmed Beckwith–Wiedemann syndrome and preauricular pits due to a duplication 22q11.21. Recent literature shows that for some CNVs, the presence of a phenotype may depend on the co-occurrence of other CNVs.15 We did not include this two-hit model in our interpretation scheme, because we feel it is, at the moment, beyond the scope of daily routine diagnostics.
To determine the value of our interpretation strategy (Figure 1), we tested it on a second cohort of 300 patients. The interpretation scheme proved to be clear, easy to follow and resulted in an efficient interpretation. In addition, during the course of the study, the following recommendations emerged.
Use of an in-house or national reference database
The use of an in-house or national database with array data obtained from controls proved to be invaluable in this study, as 68.5% and 65.3%, respectively, of the gains were concluded to be benign after comparing with this database. As the database consisted of parents who all have a child with DD/MCA, it is obviously not a completely independent control cohort. We therefore used a threshold of 1%, ensuring that this bias does not have a significant influence. The use of the Database of Genomic Variants has some shortcomings, because of the inclusion of CNVs detected by different array platforms and because some individuals may have been included who are not phenotypically normal. Nevertheless, in the first and second cohort, an additional 19% (151/797) and 16% (95/594) of the gains, respectively, were concluded to be benign, based on this database. Thus, the Database of Genomic Variants has a complementary value to our reference database, saving time-consuming literature studies.
Localise gains with FISH
The importance of FISH studies in locating the duplicated fragment was demonstrated by the intragenic gain of 253 kb in the DMD gene that appeared to be an insertion of Xp21.1 material into Xq27. We recommend that especially de novo intragenic duplications or de novo duplications with a breakpoint in a gene are located by FISH before a conclusion is drawn about their clinical relevance. For de novo duplications, in general, it is known that the majority occur in tandem, but some are the result of an insertional translocation, as recently demonstrated by Kang et al.16 Such an insertional translocation may still not have any clinical consequences if the duplicated segment is inserted in a gene desert, but it may also disrupt or otherwise influence the expression of genes at the insertion breakpoint.17 Unravelling the pathogenic nature of a submicroscopic insertional translocation requires the use of sophisticated techniques that are often not available in a routine diagnostic setting.
Set a 200-kb threshold for detecting gains in routine diagnostics
The size of a gain appeared to be a useful indicator for its clinical relevance, as such CNVs were significantly larger than benign CNVs or CNVs of unknown clinical relevance (P<0.001) (Table 3). On the basis of our data, it is acceptable to set a threshold of 200 kb for detecting clinically relevant microduplications in routine diagnostics at the moment (Table 4). Increasing the threshold results in a lower sensitivity, whereas decreasing the threshold substantially reduces the specificity.
Do not exclude a clinical relevance for gains inherited from parents
The obvious assumption that de novo CNVs most likely are pathogenic is under debate.18 We confirmed that the de novo nature of a gain does not always mean it is clinically relevant, as 94.8% (218/230) of the de novo gains in the first cohort were considered to be benign using the applied criteria. In both cohorts combined, 16 of the 26 clinically relevant gains for which the origin was known appeared to be familial.
In our study combined, 9 out of 12 gains that were associated with known microduplication syndromes and for which segregation could be esablished, were inherited. Microduplication syndromes show a highly variable penetrance between generations and they are often found to be inherited from an asymptomatic or very mildly affected parent.19, 20 If we exclude the known microduplication syndromes, still 7 of the 14 remaining clinically relevant gains with known segregation were inherited. None of these were located in a region that is known to be parentally imprinted. Five, however, involved the X chromosome in two girls and three boys, and in all three boys, these were maternally inherited. For example, both the Xq28 gains in severely affected boys were inherited from an asymptomatic mother, most likely because of X inactivation.21 Thus, the preponderance of familial clinically relevant gains in our study might be explained by the known microduplication syndromes with incomplete penetrance and the maternally inherited gains involving the X chromosome. What is important is that our results emphasise that a parental origin does not exclude clinical relevance.
CONCLUSION
We have developed guidelines for interpreting copy number gains in routine diagnostics. These guidelines proved to be clear, easy to follow and resulted in an efficient interpretation. In contrast to mode of inheritance, the minimum size of a gain was concluded to be a useful indicator for its clinical relevance.
References
Stankiewicz P, Beaudet AL : Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev 2007; 17: 182–192.
Iafrate AJ, Feuk L, Rivera MN et al: Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Sebat J, Lakshmi B, Troge J et al: Large-scale copy number polymorphism in the human genome. Science 2004; 305: 525–528.
Schinzel A : Catalogue of Unbalanced Chromosome Aberrations In Man, 2nd ed 2001, Berlin: New York, de Gruyter; p 33.
Bisgaard AM, Kirchhoff M, Nielsen JE et al: Transmitted cytogenetic abnormalities in patients with mental retardation: pathogenic or normal variants? Eur J Med Genet 2007; 50: 243–255.
de Ravel TJ, Balikova I, Thienpont B et al: Molecular karyotyping of patients with MCA/MR: the blurred boundary between normal and pathogenic variation. Cytogenet Genome Res 2006; 115: 225–230.
Mencarelli MA : Private inherited microdeletion/microduplications: implications in clinical practice. Eur J Med Genet 2008; 51: 409–416.
Koolen DA, Pfundt R, de Leeuw N et al: Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat 2009; 30: 283–292.
Gijsbers AC, Lew JY, Bosch CA et al: A new diagnostic workflow for patients with mental retardation and/or multiple congenital abnormalities: test arrays first. Eur J Hum Genet 2009; 17: 1394–1402.
Buysse K, Delle Chiaie B, Van Coster R et al: Challenges for CNV interpretation in clinical molecular karyotyping: lessons learned from a 1001 sample experience. Eur J Med Genet 2009; 52: 398–403.
Bruno DL, Ganesamoorthy D, Schoumans J et al: Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J Med Genet 2009; 46: 123–131.
Lee C, Iafrate AJ, Brothman AR : Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet 2007; 39: S48–S54.
Stankiewicz P, Pursley AN, Cheung SW : Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am J Med Genet 2010; 152A: 1089–1100.
Di Giacomo MC, Cesarano C, Bukvic N et al: Duplication of 9p11.2-p13.1: a benign cytogenetic variant. Prenat Diagn 2004; 24: 619–622.
Girirajan S, Rosenfeld JA, Cooper GM et al: A recurrent 16p12.1 microdeletion supports a two hit model for severe developmental delay. Nat Genet 2010; 42: 203–209.
Kang SL, Shaw C, Ou Z et al: Insertional translocations detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am J Med Genet 2010; 152A: 1111–1126.
Neill NJ, Ballif BC, Lamb AN et al: Recurrence, submicroscopic complexity, and potential clinical relevance of copy gains detected by array CGH that are show to be unbalanced insertions by FISH. Genome Res 2011; 21: 535–544.
Vermeesch JR, Balikova I, Schrander-Stumpel C et al: The causality of de novo copy number variants is overestimated. Eur J Hum Genet 2011; e-pub ahead of print 18 May 2011; doi:10.1038/ejhg.2011.83.
Mefford HC, Sharp AJ, Baker C et al: Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 2008; 359: 1685–1699.
Ou Z, Berg JS, Yonath H et al: Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med 2008; 10: 267–277.
Van Esch H, Bauters M, Ignatius J et al: Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–453.
Acknowledgements
We thank Jackie Senior for editorial advice.
WEB RESOURCES
Database of small supernumerary marker chromosomes, http://www.med.uni-jena.de/fish/sSMC/00START.htm. Database of Genomic Variants, http://projects.tcag.ca/variation. Decipher, http://decipher.sanger.ac.uk. Embase, http://www.embase.com/. European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations, http://www.ecaruca.net. Genatlas, http://genatlas.medecine.univ-paris5.fr. PubMed, http://www.ncbi.nlm.nih.gov/pubmed/. University of California-Santa Cruz Human Genome Browser, http://genome.ucsc.edu/.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Hanemaaijer, N., Sikkema-Raddatz, B., van der Vries, G. et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet 20, 161–165 (2012). https://doi.org/10.1038/ejhg.2011.174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.174
Keywords
This article is cited by
-
Novel Germline TET2 Mutations in Two Unrelated Patients with Autoimmune Lymphoproliferative Syndrome-Like Phenotype and Hematologic Malignancy
Journal of Clinical Immunology (2023)
-
16p13.11 microdeletion/microduplication in fetuses: investigation of associated ultrasound phenotypes, genetic anomalies, and pregnancy outcome follow-up
BMC Pregnancy and Childbirth (2022)
-
Evaluation of chromosomal abnormalities and copy number variations in fetuses with ultrasonic soft markers
BMC Medical Genomics (2021)
-
Clinical and genetic findings in patients with congenital cataract and heart diseases
Orphanet Journal of Rare Diseases (2021)
-
Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders
npj Genomic Medicine (2021)
