
Abstract
The identification of ancestral admixture proportions for human DNA samples has recently had success in forensic cases. Current methods infer admixture proportions for the target sample, but not for their parents, which provides an additional layer of information that may aid certain forensic investigations. We describe new maximum likelihood methods (LEAPFrOG and LEAPFrOG Expectation Maximisation), for inferring both an individual’s admixture proportions and the admixture proportions possessed by the unobserved parents, with respect to two or more source populations, using single-nucleotide polymorphism data typed only in the target individual. This is achieved by examining the increase in heterozygosity in the offspring of parents who are from different populations or who represent different mixtures from a number of source populations. We validated the methods via simulation; combining chromosomes from different Hapmap Phase III population samples to emulate first-generation admixture. Performance was strong for individuals with mixed African/European (YRI/CEU) ancestry, but poor for mixed Japanese/Chinese (JPT/CHB) ancestry, reflecting the difficulty in distinguishing closely related source populations. A total of 11 African-American trios were used to compare the parental admixture inferred from their own genotypes against that inferred purely from their offspring genotypes. We examined the performance of 34 ancestry informative markers from a multiplex kit for ancestry inference. Simulations showed that estimates were unreliable when parents had similar admixture, suggesting more markers are needed. Our results demonstrate that ancestral backgrounds of case samples and their parents are obtainable to aid in forensic investigations, provided that high-throughput methods are adopted by the forensic community.
Similar content being viewed by others

Introduction
Recent examples of forensic investigations inferring the genetic ancestry of DNA samples include the 11-M Madrid Bombing case1, 2 and Operation Minstead in the United Kingdom.3 Both used small panels of ancestry informative markers (AIMs) made up of single-nucleotide polymorphisms (SNPs) or short tandem repeats (STRs) that are highly differentiated in allele frequency between human populations. These are suitable for forensic work where the amount of sample DNA is typically restricted. Currently, information about maternal and paternal ancestry is derived from the mitochondrial and Y haplotypes. These can be assigned to populations of origin, but represent a small fraction of the full genome, and so cannot be assumed to be representative of all the disparate ancestries that make up a single individual.4 Whole-genome amplification, where PCR is performed using randomly generated primers, may facilitate the collection of genome-wide data from forensic samples,5 and the nascent single-molecule sequencing technologies offer similar opportunities.
We describe a statistical method, LEAPFrOG (Likelihood Estimation of Admixture in Parents From Offspring Genotypes), for quantifying the admixture proportions of a sample’s unobserved parents in addition to their personal admixture; designed to be computationally tractable over large numbers of autosomal SNPs. This parental admixture information could cast additional light on the identity of case samples, which is not possible using existing methods. We also present an alternative approach, LEAPFrOG Expectation Maximisation (EM), which is more appropriate when reliable phased data are available, for example from single-molecule sequencing. We demonstrate that the methods give useful predictions of parental admixture using genome-wide SNP platforms for divergent source populations such as those originating from different continents.
Methods
When an offspring descends from parents with different ancestries, a departure from Hardy–Weinberg equilibrium (HWE) is induced in all autosomal loci that one might genotype in the offspring. To take an extreme case, if the parents are 100% divergent, then parent 1 will be homozygous AA, parent 2 will be homozygous BB and the offspring will be heterozygous AB at all autosomal SNP loci (arbitrary allele coding), representing a severe departure from HWE. Here we leverage this type of Wahlund effect6 to infer the genetic make-up of the unobserved parents. We allow each parent to be a genetic mixture of a number of different source populations. We then estimate both the parental admixture proportions and the degree of genetic divergence between them, based only on SNP data obtained from a single offspring, together with data from a reference set of source populations.
We take a maximum likelihood estimation approach, based on the probability of seeing the observed genotypes conditional on the unknown admixture proportions in the target (offspring) individual and parental divergences in admixture. These are governed by the parameters m1…J-1 and D1…J-1, respectively, where J is the number of source populations and mJ and DJ are constrained by the other parameters. mj is the proportional contribution (admixture proportion) of source population j in the genome of the target individual, and by definition is made up of equal contributions from the genomes of the two parents. A1j and A2j are the proportional contributions of source population j in parents 1 and 2, respectively, and are determined by the underlying parental divergence parameter Dj as described by Equations 13 and 14.
For LEAPFrOG, maximum likelihood estimates are found using the first derivatives of the likelihood equations presented below. If phased data are available, we use an iterative EM algorithm,7 LEAPFrOG EM, which assigns chromosomes probabilistically to the most likely parent of origin in the expectation step, and then optimises parental admixture proportions conditional on these probabilities in the maximisation step. Details of both LEAPFrOG and LEAPFrOG EM are as follows.
LEAPFrOG
The Wahlund principle describes the departure of genotype frequencies from HWE in the first generation after divergent populations are fused. It follows that the probabilities of observing the three possible genotypes of a biallelic polymorphism are



where  and
and  are the average major and minor allele frequencies, respectively, and w is the magnitude of the departure, which is dependant on the divergence of the populations. A single offspring can be modelled as the product of an admixture event between two equal-sized ‘populations’, representing the populations from which parent 1 and 2, respectively, are drawn. If P1 and P2 are the major allele frequencies in these two populations, it can be shown that
are the average major and minor allele frequencies, respectively, and w is the magnitude of the departure, which is dependant on the divergence of the populations. A single offspring can be modelled as the product of an admixture event between two equal-sized ‘populations’, representing the populations from which parent 1 and 2, respectively, are drawn. If P1 and P2 are the major allele frequencies in these two populations, it can be shown that

We now expand the model to allow both parental ‘populations’ to be admixtures of J distinct ‘source’ populations. We let pj and qj represent, respectively, the major and minor allele frequency in population j, mj the average contribution of population j across the two parental ‘populations’ (and hence also the contribution of population j to the offspring’s genome), and Dj the degree of divergence between the two parental ‘populations’, such that Djmj and (1−Dj)mj are the weights for population j in parents 1 and 2, respectively. Expanding Equation 4:

All values of Dj and mj are constrained to be between 0 and 1. Each parent transmits half of its genetic material to the offspring, so  and
and  must be equal to 0.5. These terms are therefore multiplied by 2 in Equation 5 to obtain the allele frequencies in the parental ‘populations’.
must be equal to 0.5. These terms are therefore multiplied by 2 in Equation 5 to obtain the allele frequencies in the parental ‘populations’.
As all offspring and parental admixture proportions must sum to 1, we only estimate J−1 m and J−1 D parameters directly. DJ and mJ are replaced in the model as follows:


Furthermore, in Equation 5, there are limits on the values that Dj can take depending on mj. For example, if mj exceeds 0.5, Dj cannot be 1 as this implies that one parent is transmitting more genetic material to the offspring than the other. Therefore, we rearrange so that Dj governs the amount of possible admixture, for population j, originating from one parent. After some algebra, we define

where W(D1...J−1, m1...J−1) is equal to (P1−P2)2/4 as described in Equation 4 and I is an indicator variable, which takes the value of 1 if the condition in the parentheses are met and 0 otherwise. We use Equation 8 to measure the magnitude of the Wahlund effect, and rewrite Equations 1, 2, 3 as



Estimates for all m and D parameters are determined by maximising the following likelihood function using gradient optimisation (partial derivatives not shown):

where N denotes the number of SNPs, i is the SNP identifier and Xi is an indicator vector with three indices corresponding to the three genotypes, which takes the value 1 if that genotype is observed or 0 otherwise. SE for the parameter estimates are obtained from the square root of the inverted Hessian matrix of second-order partial derivatives, which are calculated numerically during optimisation.
We apply an additional constraint to prevent D1 taking values <0.5. The problem then becomes asymmetrical and a parameter nonidentifiability issue is circumvented. The admixture proportions for parents 1 and 2 (A1j and A2j, respectively) can be easily calculated from the estimates of Dj and mj:


LEAPFrOG EM
We begin with phased parental chromosomes, where we know that alleles on the same chromosome must come from the same parent, but alleles on separate chromosomes can come from different parents. The objective is to separate the chromosomes into two groups, each representing a parent, and estimate admixture proportions for both. We assume that the admixture pattern for each transmitted chromosome is the same as the admixture pattern for the full parental genome.
Expectation step
We consider two alternatives H=1 and H=2 for each chromosome, which are the two ways of allocating the homologous chromosomes to parent 1 and 2 respectively. Denote p(H=1) and p(H=2) as the likelihoods under these respective hypotheses. We calculate the responsibility  for the cth chromosome as the ratio p(H=1)/(p(H=1)+p(H=2)). This can also be written as
for the cth chromosome as the ratio p(H=1)/(p(H=1)+p(H=2)). This can also be written as

where c is the chromosome pair identifier and


where t denotes the iteration (initially 1), xc1 and xc2 are the data for chromosomes 1 and 2 in the homologous pair and μ1and μ2 are vectors of population proportions in parents 1 and 2 respectively. It follows that



where Xi1 and Xi2 are indicator values for each chromosome in the homologous pair, which are 1 if the major allele for SNP i is present on that chromosome or 0 otherwise, and Nc is the number of SNPs on the chromosome. l0c is calculated as

Maximisation step
Here we choose the parameter vectors  and
and  using gradient optimisation as follows:
using gradient optimisation as follows:

Where C is the number of chromosome pairs. Again, we only estimate μ for the first J−1 populations:

The full process is iterated over t until the change in parameters becomes negligible. We skip the first expectation step, assigning responsibilities ( ) of 0.5 to all homologous chromosomal pairs save the first, which we set to 1. This is helpful because μ1 and μ2 are designated arbitrarily to the parents, and thus it avoids a nonidentifiability issue. SEs are calculated using the Hessian matrix produced by gradient optimisation during the maximisation step.
) of 0.5 to all homologous chromosomal pairs save the first, which we set to 1. This is helpful because μ1 and μ2 are designated arbitrarily to the parents, and thus it avoids a nonidentifiability issue. SEs are calculated using the Hessian matrix produced by gradient optimisation during the maximisation step.
Equation 21 involves calculating the product of many probabilities, which results in extremely small numbers. To achieve this, we use a python script which can operate with arbitrary floating point precision.
The computation time is largely dependant on the ancestral similarity of the parents. Convergence for well-diverged parents typically occurs at three iterations, but can take longer when divergence is low.
Application to real and simulated data
To validate the methods, we first took 100 phased haploid genomes from unrelated individuals in Hapmap Phase III CEU (www.hapmap.org) and combined them with 100 unrelated YRI haploid genomes to create synthetic first-generation admixed individuals, which were analysed using both LEAPFrOG and LEAPFrOG EM. The source populations were East Asia, Europe and West Africa, represented by 170 JPT+CHB, 112 CEU and 113 YRI individuals. SNPs with genotyping rates of <98% across the populations were removed, leaving 994 200 SNPs at the analysis stage. As the parents of the synthetic individuals come from the same source data sets, both of these were excluded before calculating source-population allele frequencies for use in maximum likelihood estimation. We also analysed 100 first-generation admixed individuals with parents from Japan and China (86 JPT and 84 CHB individuals), simulated using the same SNPs.
We examined African, East Asian and European parental admixture in 83 African-American (ASW) individuals from the Hapmap phase III data set. The analysis was performed on 1 078 914 autosomal SNPs (1 011 119 when using phased data for LEAPFrOG EM), which exceeded a 98% genotyping rate. Real genome-wide SNP data contain dependencies between SNPs due to linkage disequilibrium (LD), which violates an assumption made by the LEAPFrOG methods. Although it is possible to ‘prune’ markers from the data in order to reach some nominal low value of LD, here we are more interested in maximising the accuracy of the point estimates, at the expense of some underestimation of the SEs. We analysed the effect of using independent SNPs by comparing LEAPFrOG results for the ASW individuals with those for an ‘LD pruned’ version of the data described above, where 53 783 SNPs with pairwise r2 <0.1 were retained. The LD pruned data was also used to compare LEAPFrOG predictions of offspring admixture proportions for the ASW individuals with those from ADMIXTURE,8 an alternative method, which estimates admixture proportions in the target individual only.
Phillips and colleagues designed a panel of 34 ancestrally informative SNPs from Hapmap and the 1000 Genomes Project.2, 9 With these, we predicted African and European admixture proportions in simulated individuals, using 225 Africans from Nigeria, Senegal, The Democratic Republic of Congo, Kenya, Somalia, The Central African Republic, Mozambique, South Africa and Namibia, and 278 Europeans from Spain, France, Scotland, Italy, Denmark and Russia. These genotypes were collated by the SNPforID consortium (http://www.snpforid.org). Two triallelic SNPs were removed, being incompatible with the likelihood method, plus a biallelic SNP, which has poor genotyping rates in casework,1 leaving 31 for analysis. A total of 100 simulations of both first-generation admixture and 0% parental divergence (each parent having 50/50 African/European ancestry) were performed.
To explore the relationship between population differentiation and admixture predictability, we simulated two ancestral populations under a Balding–Nichols model10 and then generated 100 individuals with first-generation admixture. This simulation was then repeated with 0% parental divergence, where the parents of the individuals are both 50% admixed between the two Balding–Nichols populations. Allele frequencies from the two populations were used to predict admixture and parental admixture from the offspring genotypes, and Fst between the ancestral populations was varied from 0 to 0.1 in increments of 0.005. A further simulation was performed where the true allele frequency, derived from the Balding–Nichols model, was replaced with one calculated from 400 simulated individuals (200 from each population) to approximate the sampling error that we would expect for real data sets. Simulations were for 80 000 independent SNPs, roughly equivalent to the number of independent regions in the human autosomal genome.
Further Balding–Nichols simulations were performed to assess reliability across various parental divergences and number of SNPs. Two populations were simulated with an Fst of 0.15, and the target sample had 50% admixture from each. We recorded the mean squared error between the estimated and simulated parameters across 1000 simulated individuals, under various parental admixture proportions (mean across parents always 0.5/0.5) and numbers of SNPs. We also calculated the proportion of times that the 95% confidence intervals for the estimates covered the simulated parameter value (coverage probability).
Results
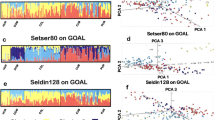
With genome-wide SNP data, the LEAPFrOG method gave accurate prediction results for YRI/CEU synthetic first-generation admixture (Figure 1a, Supplementary Table 2), making it possible to reliably infer admixture proportions in both the unobserved parents and the genotyped target individual. For all the simulations, East Asian ancestry was correctly found to be low (mean 0.006), and African and European admixture proportions approximately equal (mean 0.495, 0.499), but highly divergent in terms parental origin. Results from the LEAPFrOG EM method were similar (results not shown).

(a, b) LEAPFrOG-estimated admixture proportions in synthetic individuals with first-generation admixture. Each vertical bar shows the admixture proportions for one individual, and the order of bars in each panel is consistent with the parental/offspring relationships. (a) 100 individuals with European/West African (CEU/YRI) admixture using 994,200 SNPs from Hapmap phase III. (b) 100 individuals with Japanese/Chinese (JPT/CHB) admixture using the same SNPs. (c) LEAPFrOG parameter estimation for 83 African-American (ASW) individuals using 1,078,914 SNPs from Hapmap phase III. For full model estimates see Supplementary Tables 2–4.
JPT/CHB first-generation admixture was harder to predict (Figure 1b, Supplementary Table 3). Target individual admixture proportions were correctly distributed around 0.5 but variation in these estimates was greater than in the YRI/CEU results. Parental predictions were highly variable with LEAPFrOG, although LEAPFrOG EM returned more consistent estimates, with mean CHB admixture of 0.767 (0.766–0.768) in the first parent and 0.224 (0.223–0.225) in the second (full results not shown).
The African-American ASW individuals (Figure 1c, Supplementary Table 4) demonstrated admixture proportions approximately in line with prior expectations11 (mean African admixture of 0.773 in target individuals), and a range of parental divergence. The mean difference in African admixture between parents was 0.118, and this increased to 0.172 when using LEAPFrOG EM (Supplementary Figure 1).
The ASW set contains 11 trio sets, allowing a comparison to be made between the parental admixture proportions predicted from the target and parent genotypes (Figure 2, Supplementary Figure 2). LEAPFrOG EM results had marginally lower variance around the expected (full concordance between estimates) than LEAPFrOG (0.004 versus 0.005).

Comparison of LEAPFrOG parental admixture proportions estimated from offspring genotypes and directly from parental genotypes in 11 African-American trios genotyped at 1,078,914 SNPs.
Comparing LEAPFrOG results for the LD pruned and unpruned ASW data set showed that African admixture proportions for the offspring were on average 2.1% (1.9–2.1) higher for the latter (Supplementary Figure 4). We also estimated African admixture proportions using the ADMIXTURE program8 and found that they were on average 1.7% (0.69–1.71) higher than when using LEAPFrOG (Supplementary Figure 5). These deviations are relatively small, and we consider the reasons why they occur in the Discussion.
We investigated LEAPFrOG prediction results from 31 AIM SNPs for simulated admixture between Africa and Europe (Figure 3, Supplementary Tables 5 and 6). Results were quite accurate for first-generation admixture (parents either fully ‘African’ or ‘European’) but highly variable when both parents were simulated with identical admixture proportions.

LEAPFrOG estimates of offspring and parental admixture proportions from 31 ancestrally informative markers, for (a) 100 simulated individuals with first-generation admixture between Africa and Europe; (b) 100 simulated individuals whose parents have 50% African and 50% European admixture. Each bar shows the admixture proportions for one individual. For full model estimates see Supplementary Tables 5 and 6.
The Balding–Nichols simulation results across various levels of population divergence (Supplementary Figure 3) showed that first-generation admixture could be accurately inferred using LEAPFrOG for Fst greater than approximately 0.02. Non-divergent parental admixture is more difficult to predict, and even distantly related populations (Fst=0.1) fail to provide mean admixture proportions of 0.50 in each parent. The simulations across various parental divergences and numbers of SNPs (Table 1) show that 60 000 SNPs provide excellent accuracy in terms of mean squared error for parental divergence, but good results can be obtained with fewer markers, particularly at higher levels of divergence. Confidence interval coverage was poor at the boundary values but good otherwise, with the true parameter value within the 95% confidence intervals in approximately 95% of simulations. Parameter estimates and coverage for offspring admixture were highly accurate (Supplementary Table 1), though confidence intervals appeared slightly conservative when parental divergence was high.
Discussion
The identification of ethnic or geographic origins has proved useful in some forensic investigations.1 Our method takes this one step further by inferring the origins of the unknown DNA sample’s parents, something not attempted by any of the existing methods for admixture estimation (eg Frappe,12 Structure13 and ADMIXTURE8). Different admixture proportions from multiple source populations are allowed in either parent, thus allowing for complex admixture patterns rather than ‘all-or-nothing’ classification to a single-source population.
Our results demonstrate the efficacy of LEAPFrOG and LEAPFrOG EM for well-differentiated populations, such as Africa and Europe, using genome-wide SNP data; particularly when parents are highly divergent for ancestry. LEAPFrOG EM gave more consistent parental admixture estimates than LEAPFrOG, underlining the potential utility of pre-phased data. However, neither method was able to accurately identify first-generation admixture events between China and Japan, showing that parental ancestry prediction may be hard to achieve when the source populations are closely related.
Comparison of parental admixture predictions from their observed genotypes with those from the genotypes of their offspring, using the 11 African-American trios, suggest a reasonable ability to estimate parental admixture proportions using offspring data alone (Figure 2, Supplementary Figure 2). LEAPFrOG estimates for all 83 African-Americans reveal extensive diversity in both offspring admixture and parental admixture divergence. Three individuals were predicted to have a considerable East Asian component (Figure 1c), the most likely explanation of which is Native American ancestry, as this population would be most similar to JPT/CHB among the source groups. LEAPFrOG EM detected greater African admixture divergence between parents than LEAPFrOG, demonstrating the advantages conferred by phase information when the admixture is complex. However, phased data in these applications should be treated with care, as existing statistical phasing methods assume a simple population history at odds with the complex admixture patterns being investigated here. This obstacle should be removed by the advent of experimentally determined phase from single-molecule sequencing technology.14
Balding–Nichols simulations indicated that accurate LEAPFrOG prediction of first-generation admixture with genome-wide microarray panels may be possible with Fst as low as 0.01, the same as between Latvia and Spain,15 two widely spaced European countries. The sampling error (n=200 for each source population) had a considerable effect on prediction when Fst <0.03, suggesting that our results for JPT/CHB (combined n=170) admixture might have been more accurate if larger samples were available. The Fst between these populations is 0.0069.16
Our simulations showed that the mean squared error for the D parameters depends largely on the number of SNPs and extent of parental divergence, with low divergence requiring more markers to accurately estimate. The confidence interval coverage (the proportion of times the confidence intervals capture the simulated parameter value) is poor at the parameter boundaries but otherwise good. D1 cannot take values <0.5 or >1 (see Methods), so the distribution of estimates is not normal here, which explains why the confidence intervals do not behave as expected. The anti-conservative confidence intervals, which occur when parental divergence is 0%, are the result of a bimodal distribution for the parameter estimates, which disappears when an extremely high number of independent SNPs (500 000) is simulated. Both mean squared error and confidence interval coverage are generally excellent for m, the offspring admixture proportions (Supplementary Table 1), although confidence intervals appear somewhat conservative when parental divergence is maximal. We have also implemented a Bayesian model with user customisable Dirichlet priors for parental admixture, which can be used to generate credible intervals, but this method does not scale to full genome-wide data.
Our methods do not allow one to determine which of the two inferred parental admixture patterns belongs to the mother and which to the father. However, it may be possible to cross-compare with mtDNA or Y-chromosome data to shed light on this. Furthermore, if the DNA sample is male then the X-chromosome is guaranteed to come from the mother, and so separate analysis of this chromosome may allow one to distinguish the maternal and paternal admixture pattern.
Both LEAPFrOG and LEAPFrOG EM assume independence between markers, in contrast with real genome-wide data, which contain dependencies among markers due to LD. Using all available data, while violating the assumption of independence, provides the best point estimates at the expense of underreporting the width of the confidence intervals. Users should decide which of these quantities they consider to be more important and filter the data accordingly (for example by ‘pruning’ the data for LD). Using African-American individuals, we demonstrated that the average discrepancy between African admixture estimates from LD ‘pruned’ and ‘unpruned’ data was 2.1%. The systematic difference is because the uncertainty in the largest admixture proportion is predominantly one-sided, as estimates cannot be >1.
The innovative aspect of LEAPFrOG lies in its ability to predict admixture in the unobserved parents of a target DNA sample, but it is nevertheless of interest to compare the target individual’s estimated admixture proportions with those obtained with an existing method such as ADMIXTURE.8 Our comparison revealed small but consistent discrepancies, which we attribute to differences in the approach taken to modelling the underlying source populations. LEAPFrOG assumes that the allele frequencies in these are known, whereas ADMIXTURE takes a clustering approach in which they are re-estimated based on the admixture across all individuals. LEAPFrOG could be extended to treat the source populations in the same way as ADMIXTURE. Although this could be advantageous in some circumstances, we found only small differences in the admixture estimates when both methods were applied to African-American individuals.
Prediction of parental admixture proportions was not accurate using a 34-AIM (31 after quality control) PCR multiplex panel. The increase in heterozygosity one sees after admixture events is subtle and requires large numbers of markers to exploit. The ability to make inferences about parental ancestry therefore seems limited to situations where genome-wide data, for example from SNP microarrays or whole-genome sequencing, are available to investigators. It should be possible to extend the methods to facilitate the analysis of STRs, but at present they are limited to biallelic markers. Although the ability to collect genome-wide SNP data for the target individual depends largely on the quantity and quality of DNA, there must also be a sufficiently large intersection with SNPs genotyped at the desired source populations. Furthermore, many individuals are needed at each population to accurately estimate allele frequencies. Most applications will therefore be limited to cases where genome-wide data is publicly available for these populations. The best current resources are the International Hapmap and 1000 Genomes projects.
Our results, as examples of the detailed information attainable from DNA samples, advocate the integration of high-throughput technology into forensic casework. The methods described in this paper are implemented as an R package, LEAPFrOG, available from CRAN (http://cran.r-project.org/web/packages/LEAPFrOG). This includes a Bayesian method not presented in detail here.
References
Phillips C, Prieto L, Fondevila M et al. Ancestry analysis in the 11-M Madrid bomb attack investigation. PLoS One 2009; 4: e6583.
Phillips C, Salas A, Sanchez JJ et al. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs. Forensic Sci Int Genet 2007; 1: 273–280.
Jacobson P : Investigation: Stalker in the suburbs. The Sunday Times 2005.
King TE, Parkin EJ, Swinfield G et al. Africans in Yorkshire? The deepest-rooting clade of the Y phylogeny within an English genealogy. Eur J Hum Genet 2007; 15: 288–293.
Schneider PM, Balogh K, Naveran N et al. Whole genome amplification—the solution for a common problem in forensic casework? Int Congr Ser 2004; 1261: 24–26.
Wahlund S : Composition of populations and correlation appearances viewed in relation to the studies of inheritance. Hereditas 1928; 11: 65–106.
Dempster AP, Laird NM, Rubin DB : Maximum likelihood from incomplete data via em algorithm. J Roy Stat Soc B Met 1977; 39: 1–38.
Alexander DH, Novembre J, Lange K : Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664.
Durbin RM, Abecasis GR, Altshuler DL et al. A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Balding DJ, Nichols RA : A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 1995; 96: 3–12.
Price AL, Tandon A, Patterson N et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet 2009; 5: e1000519.
Tang H, Peng J, Wang P, Risch NJ : Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 2005; 28: 289–301.
Pritchard JK, Stephens M, Donnelly P : Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H : Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 2009; 4: 265–270.
Nelis M, Esko T, Magi R et al. Genetic structure of Europeans: a view from the North-East. PLoS One 2009; 4: e5472.
Heath SC, Gut IG, Brennan P et al. Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet 2008; 16: 1413–1429.
Acknowledgements
We would like to thank the Biotechnology and Biological Sciences Research Council (BBSRC) for funding. We also thank Chris Phillips, Barbara Daniel, Denise Syndercombe Court and members of the Statistical Genetics Unit for advice and discussion.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Crouch, D., Weale, M. Inferring separate parental admixture components in unknown DNA samples using autosomal SNPs. Eur J Hum Genet 20, 1283–1289 (2012). https://doi.org/10.1038/ejhg.2012.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.134
