
Abstract
Short stature homeobox-containing gene, MIM 312865 (SHOX) is located within the pseudoautosomal region 1 (PAR1) of the sex chromosomes. Mutations in SHOX or its downstream transcriptional regulatory elements represent the underlying molecular defect in ∼60% of Léri-Weill dyschondrosteosis (LWD) and ∼5–15% of idiopathic short stature (ISS) patients. Recently, three novel enhancer elements have been identified upstream of SHOX but to date, no PAR1 deletions upstream of SHOX have been observed that only encompass these enhancers in LWD or ISS patients. We set out to search for genetic alterations of the upstream SHOX regulatory elements in 63 LWD and 100 ISS patients with no known alteration in SHOX or the downstream enhancer regions using a specifically designed MLPA assay, which covers the PAR1 upstream of SHOX. An upstream SHOX deletion was identified in an ISS proband and her affected father. The deletion was confirmed and delimited by array-CGH, to extend ∼286 kb. The deletion included two of the upstream SHOX enhancers without affecting SHOX. The 13.3-year-old proband had proportionate short stature with normal GH and IGF-I levels. In conclusion, we have identified the first PAR1 deletion encompassing only the upstream SHOX transcription regulatory elements in a family with ISS. The loss of these elements may result in SHOX haploinsufficiency because of decreased SHOX transcription. Therefore, this upstream region should be included in the routine analysis of PAR1 in patients with LWD, LMD and ISS.
Similar content being viewed by others

Introduction
Short stature homeobox-containing gene, MIM 312865, located in the pseudoautosomal region 1 (PAR1) of the X and Y chromosomes,1, 2 encodes a transcription factor implicated in skeletal development. Heterozygous mutations in SHOX or the downstream enhancer elements have been associated with ∼60% of Léri-Weill dyschondrosteosis (LWD, MIM 127300) and 5–15% of idiopathic short stature (ISS, MIM 300582) cases.3, 4, 5 LWD is a skeletal dysplasia, characterized by disproportionate short stature, mesomelic limb shortening and the Madelung deformity. ISS is a condition defined as a height below −2 SDS in the absence of known specific causative disorders.6 Langer mesomelic dysplasia (LMD, MIM 249700) represents a more severe clinical form with a disproportionate short stature, due to mesomelic and rhizomelic limb shortening abnormality of the humeral head, angulation of the radial shaft, carpal distortion, short femoral neck, and hypoplastic or absent proximal half of the fibula. Homozygous or compound heterozygous mutations of SHOX and/or its downstream enhancers are associated with 75% of LMD cases.7, 8, 9 The molecular defect in the remaining fraction of LWD, LMD and ISS cases are unknown.
Recently, three SHOX enhancer elements have been identified in the PAR1 upstream of SHOX.10 We screened this upstream region for mutations in 63 LWD and 100 ISS patients with no known alteration in SHOX or its downstream regulatory elements. We identified one deletion that encompassed two of the three upstream enhancers in a family with ISS.
Materials and methods
Clinical patients
The study was approved by the local Ethical Committees and all participants provided informed consent for the performed studies. The LWD and ISS patient samples were referred from endocrinology and genetic clinics. LWD patients were ascertained using the inclusion criteria of the presence of the Madelung deformity and mesomelic shortening of the limbs in the proband or a direct family member. Stature was recorded and SDS were determined according to the population standards for age and gender. ISS patients with stature <−2 SDS were ascertained using the current consensus criteria.6 Peripheral blood was drawn from probands and when possible, from relevant family members. Genomic DNA was isolated from whole blood using the salt precipitation method (Qiagen Blood Core B Kit, Qiagen, Valencia, CA, USA).
A total of 63 LWD and 100 ISS patients with no known pathogenic PAR1 defect were analyzed. In all cases, the presence of alterations in the SHOX or the downstream enhancer region had been previously excluded by MLPA or microsatellite analysis and by dHPLC or high-resolution melting analysis and DNA sequencing.3, 11, 12
A panel of 340 normal individuals, obtained from the Spanish DNA bank (University of Salamanca, Salamanca, Spain), with heights within the normal range for the Spanish population for age and gender (−2 < SDS < +2), was also screened.
MLPA assay
A specific MLPA assay was designed to screen for deletions and duplications upstream of SHOX. The MLPA consists of 16 probes: 13 localized upstream of SHOX, three of which are located within the three SHOX enhancer elements and three control autosomal probes (Supplementary Table 1). This MLPA was performed using the above Salsa probe mix and the commercial reagents EK5 (MRC Holland, Amsterdam, The Netherlands). Data were analyzed as described previously.12
Array CGH
Fine-tiling Y chromosome-specific CGH arrays (Roche NimbleGen, Madison, WI, USA) were undertaken using the service provided by Imagenes, Berlin, Germany. Fluorescence intensities and log2 intensity ratios for the test versus control sample were calculated using the Signalmap software (Roche NimbleGen).
Results
A heterozygous upstream SHOX deletion was identified in one Spanish patient with ISS (Figure 1). No other alterations were identified in the studied cohort of 63 LWD and 100 ISS cases with no known SHOX or downstream enhancer alteration.

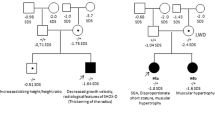
Schematic representation of the genomic location and approximate extensions of the observed upstream SHOX deletion. The approximate coordinates are according to chromosome X, NCBI assembly GRCh37 with the upstream enhancers indicated as CNE2, 3 and 5.10 The MLPA probes are indicated by grey boxes. *Two genes/loci that are included in both the commercial and the upstream MLPA assays but the binding sequences are different ▴, the presence of a heterozygous deletion. The deletion size range is indicated adjacent to each individual. The deletion detected in a patient with brachymesomelic dysplasia with Peters anomaly of the eye is also shown.14 The extension limits were determined according to the reported array and PCR data. Black solid lines indicate deleted sequences, white lines indicate the presence of two copies while hashed lines indicate non-informative areas. The diagram is not drawn to scale.
The deletion was initially detected via the deletion of a single probe included in the commercial MLPA Salsa P018 kit (MRC Holland), covering the Protein Phosphatase 2, Regulatory Subunit B-Double Prime β gene (PPP2R3B, MIM 300339), localized ∼307 kb from the telomere (probe 09333-L10292). This gene encodes a subunit of a serine/threonine phosphatase, which exerts regulatory control over DNA replication initiation.13 The deletion of this gene was thought unlikely to be the cause of the observed short stature and no further analysis was undertaken at this time point.
This patient was subsequently re-evaluated with the self-designed SHOX upstream MLPA. The upstream deletion was confirmed and further delimited (Figure 1). The deletion was further characterised using Y chromosome-specific array CGH (Figure 2), which delimited the deletion size to ∼286 kb, located ∼95 kb upstream of SHOX. The aCGH data indicated that the deletion extends from ∼124349 to ∼409949 (Y chromosome, NCBI assembly GRCh36). Despite numerous attempts we were unable to cross the deletion breakpoints, probably because of the high incidence of repetitive elements.

Characterization of the upstream SHOX deletion by fine-tiling Y chromosome aCGH (Nimblegen Y chromosome-specific array). Fluorescence log2 intensity ratios of test to reference are shown for each oligonucleotide from telomere to centromere across the short arm of the Y chromosome. Sequence coordinates are taken from the Y chromosome assembly (NCBI assembly GRCh36). The log2 ratio data are shown in the NimbleGen SignalMap data viewer and each point indicates the midpoint of a 300 bp window average, which has been calculated from 1–15 probes. The deletion extensions are indicated by the line marked with an asterisk (*) and correspond from ∼124349 to ∼409949 (Y chromosome, NCBI assembly CRCh36), which corresponds to ∼134349–439949 on the chromosome Y, NCBI assembly GRCh37 (∼184349–489949 on the chromosome X, NCBI assembly GRCh37 for cross referencing with Figure 1).
To investigate the pathogenicity of the alteration, we screened the parents and a cohort of normal height controls for the upstream deletion to assess cosegregation with the short stature. The deletion was observed in the father but absent in 340 normal height controls.
Discussion
We have identified the first PAR1 deletion that does not include SHOX but encompasses the upstream SHOX enhancer elements. We propose that the deletion is pathogenic through the loss of at least two SHOX transcriptional regulatory elements of SHOX, which may decrease SHOX transcription and its expression, resulting in SHOX haploinsufficiency. The deletion may also include additional, as yet unidentified, SHOX regulatory elements. With the current data we cannot exclude the less likely option that haploinsufficiency of the protein phosphatase 2A regulatory subunit may be the causative factor.
The upstream deletion was identified in a Spanish girl, who had attended an endocrinology clinic from the age of 11.7 years for short stature (−2.14 SDS). She was vaginally delivered at full-term with a body length of 50 cm (0.11 SDS) and body weight of 3150 g (−0.35 SDS). At the most recent examination, age 13.3 years, she had a height SDS of −2.24, Tanner stage 4, and all other anthropometric measurements were normal: arm span:height ratio 0.99 and sitting height ratio of 0.50. No mesomelic shortening or Madelung deformity was observed. GH and IGF-I levels were in the normal range. The father and mother's height were 163 cm (−2.23 SDS and 162 cm (−0.28 SDS), respectively, and no bone shortening or malformation, including the Madelung deformity was observed in either parent. The mother had her menarche at 14 years. Both parents had normal GH and IGF-I levels. The upstream deletion was also detected in the father, thus showing cosegregation with the short stature.
Only one SHOX upstream deletion has been reported previously, in a family with brachymesomelic dysplasia and Peters anomaly of the eyes.14 The mother presented a pericentric inversion of X chromosome (46, X, inv(X; p22.3q27)) and the son presented a recombinant X chromosome (46,Y, rec(X)dup(Xq)inv(X)p22.3q27) with a telomeric deletion 5′ of SHOX. Both mother and son presented with severe short stature, mesomelic shortening and multiple bone malformations, an intermediate form between LWD and LMD, plus the Peters anomaly of the eyes. The breakpoints lay 30–68 kb upstream of SHOX and close to SOX3. The authors speculated that the misexpression of both genes may occur because of positional effects. Indeed, the deletion, present in the mother and son, included two or all three of the SHOX upstream enhancers but in our opinion, the phenotype appears too severe to be solely due to this PAR1 deletion.
The high incidence of repetitive sequences throughout the PAR1 has resulted in a higher than average recombination fraction15 and subsequently a higher incidence of deletions and duplications in this region. Despite the high incidence of PAR1 deletions that include the downstream SHOX enhancers (three to five) we have only observed one deletion of the upstream SHOX enhancers using the commercial MLPA (>2000 LWD and ISS) in our studied cohort using the SHOX upstream-specific MLPA probes. The incidence of deletions in this upstream region may indeed be lower than in the downstream region, but ascertainment bias may be also occurring for two reasons, first there is a poorer probe density in this region in the commercially available SHOX and PAR1 MLPA kits, and second, the associated phenotype, as suggested by our case phenotype, may be milder and does not lead to ascertainment at genetic and endocrinology clinics.
In conclusion, we have identified the first deletion that includes two upstream SHOX enhancers in a patient with ISS. Although the incidence of deletions in this region appears to be lower than deletions of the downstream enhancers, we suggest that the screening of this upstream region should be also included in the routine PAR1 screening of LWD, LMD and ISS patients.
References
Rao E, Weiss B, Fukami M et al: Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997; 16: 54–63.
Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, Chiong W : PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet 1997; 6: 1341–1347.
Benito-Sanz S, Thomas NS, Huber C et al: A novel class of pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am J Hum Genet 2005; 77: 533–544.
Huber C, Rosilio M, Munnich A, Cormier-Daire V, the French SHOX GeNeSIS Module: High incidence of SHOX anomalies in individuals with short stature. J Med Genet 2006; 43: 735–739.
Chen J, Wildhardt G, Zhong Z et al: Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J Med Genet 2009; 46: 834–839.
Cohen P, Rogol AD, Deal CL et al, on behalf of the 2007 ISS Consensus Workshop participants: Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 2008; 93: 4210–4217.
Zinn AR, Wei F, Zhang L et al: Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 2002; 110: 158–163.
Campos-Barros A, Benito-Sanz S, Ross JL, Zinn AR, Heath KE : Compound heterozygosity of SHOX-encompassing and downstream PAR1 deletions results in Langer mesomelic dysplasia (LMD). Am J Med Genet A 2007; 143A: 933–938.
Bertorelli R, Capone L, Ambrosetti F et al: The homozygous deletion of the 3′ enhancer of the SHOX gene causes Langer mesomelic dysplasia. Clin Genet 2007; 72: 490–491.
Durand C, Bangs F, Signolet J, Decker E, Tickle C, Rappold G : Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur J Hum Genet 2010; 18: 527–532.
Benito-Sanz S, del Blanco DG, Aza-Carmona M et al: PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Léri-Weill dyschondrosteosis (LWD) probands. Hum Mutat 2006; 27: 1062.
Benito-Sanz S, Barroso E, Heine-Suñer D et al: Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab 2011; 96: E404–E412.
Yan Z, Fedorov SA, Mumby MC, Williams RS : PR48, a novel regulatory subunit of protein phosphatase 2A, interacts with CDC6 and modulates DNA replication in human cells. Mol Cell Biol 2000; 20: 1021–1029.
Bleyl SB, Byrne JL, South ST et al: Brachymesomelic dysplasia with Peters anomaly of the eye results from disruptions of the X chromosome near the SHOX and SOX3 genes. Am J Med Genet A 2007; 143A: 2785–2795.
May CA, Shone AC, Kalydjieva L, Sajantila A, Jeffreys AJ : Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat Genet 2002; 31: 272–275.
Acknowledgements
This work was supported by grants from the ‘Fondo de Investigación Sanitaria’ (FIS PI08/90270), ‘Ministerio de Ciencia e Innovación’ (MICINN SAF2009-08230). SBS is a recipient of a CIBERER postdoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Benito-Sanz, S., Aza-Carmona, M., Rodríguez-Estevez, A. et al. Identification of the first PAR1 deletion encompassing upstream SHOX enhancers in a family with idiopathic short stature. Eur J Hum Genet 20, 125–127 (2012). https://doi.org/10.1038/ejhg.2011.210
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.210
Keywords
This article is cited by
-
Rare dosage abnormalities flanking the SHOX gene
Egyptian Journal of Medical Human Genetics (2021)
-
Variants in the 5′UTR reduce SHOX expression and contribute to SHOX haploinsufficiency
European Journal of Human Genetics (2021)
-
Identification of a limb enhancer that is removed by pathogenic deletions downstream of the SHOX gene
Scientific Reports (2018)
-
SHOX haploinsufficiency presenting with isolated short long bones in the second and third trimester
European Journal of Human Genetics (2018)
-
Identification of 15 novel partial SHOX deletions and 13 partial duplications, and a review of the literature reveals intron 3 to be a hotspot region
Journal of Human Genetics (2017)
