
Abstract
Preeclampsia (PE) is a serious complication of pregnancy, which is highly correlated with later life cardiovascular disease (CVD). Many risk factors are common for both diseases, but the contribution of shared genes remains to be determined. In this study, we used an integrative strategy to assess lipid traits as risk factors for PE and CVD by whole genome transcriptional profiling performed on Norwegian decidua basalis tissues (N=95) from preeclamptic and normal pregnancies and on blood lymphocytes (N=1240) from the San Antonio Family Heart Study (SAFHS). Among 222 genes that were differentially expressed (false discovery rate (FDR) P-value <0.05) between the PE, cases and controls, we found one gene, ACOX2 (acyl-coenzyme A oxidase 2, branched chain), that was downregulated in PE whose transcription was also inversely correlated with triglyceride levels (P=5.6 × 10−7; FDR P-value=0.0002) in SAFHS. We further report associations between SNPs in the ACOX2 gene and the transcription level (P-value=0.0045) of the gene, as well as with triglyceride levels (P-value=0.0051). ACOX2 is involved in bile acid production, a process that has been associated with both oxidative stress and regulation of triglyceride levels. Oxidative stress and increased triglyceride levels are known risk factors for CVD and both have also been associated with PE. Our results suggest that downregulation of ACOX2 is a shared risk factor for PE and CVD.
Similar content being viewed by others


INTRODUCTION
Preeclampsia (PE) is a pregnancy-specific disorder. With 50 000 deaths annually and over four million incidences, PE is one of the leading causes of maternal and perinatal mortality worldwide.1 Even though PE is a common disorder, which complicates about 3–5% of all pregnancies, there is still no effective treatment available except delivery. Consequently, PE accounts for approximately 20% of all preterm births.2 The pathogenesis of PE is not understood clearly. It might include endothelial dysfunction and inflammation, similar to that associated with cardiovascular disease (CVD).3 Both PE and CVD also share several metabolic abnormalities,4 including increase in small, dense low-density lipoprotein and triglycerides.5 Still, years after PE, many women have increased blood pressure, insulin resistance, triglycerides, and uric acid compared to women with normal pregnancies. Pregnancy constitutes a vascular and metabolic stress and one hypothesis is that women in the risk group of developing metabolic syndrome respond to pregnancy in an abnormal way. Thus, the same predisposing factors giving rise to PE during pregnancy may later in life cause CVD. PE has also been associated with an increased risk of later life CVD.6, 7
PE is a complex disorder influenced by multiple genetic and environmental factors and their interactions. A number of environmental risk factors have been identified and the heritability has been estimated to be 0.54.8 Transcriptional profiling has been shown to be a powerful method to identify genetic variants influencing common human traits.9 Many genes have also been identified to be differentially expressed in the placenta or in decidual tissues between women with PE and women with normal pregnancies.10, 11, 12 However, it is still unknown whether such differential gene expression is causal or is secondary to the disorder.
The aim of this study was to identify shared genetic risk factors for PE and CVD by integrating genome-wide decidual transcriptomic data from preeclamptic and control pregnancies ascertained in Norway with genome-wide lymphocyte transcriptomic data, genome-wide SNP genotyping data and CVD-related lipid phenotypic data from the San Antonio family heart study (SAFHS).
MATERIALS AND METHODS
The Norwegian PE case–control study
Study groups
Pregnant women delivered by cesarean section were recruited at St Olavs University Hospital (Trondheim, Norway) and Haukeland University Hospital (Bergen, Norway) from 2002 to 2006. PE was defined as persistent hypertension (blood pressure of ≥140/90 mm Hg) plus proteinuria (≥0.3 g per day or ≥1+ according to a dipstick test) on at least two occasions, developing after 20 weeks of pregnancy.13 Cesarean section was performed on preeclamptic women on medical indications. Exclusively women with normal pregnancies and no previous history of PE were accepted in the control group. The cesarean sections on controls were carried out for reasons such as breech presentation, cephalopelvic disproportion in earlier pregnancies or maternal request. Women with previous pregnancy complications (such as PE or intrauterine growth retardation) as well as women with multiple pregnancies, pregnancies with chromosomal aberrations, fetal and placental structural abnormalities, or suspected perinatal infections were not enrolled as controls. Informed consent was obtained from all participants before collection of decidual samples and the study was approved by the Norwegian Regional Committee for Medical Research Ethics (REK 106-03).
Transcriptional profiling
Transcriptional profiling was performed on decidua basalis tissues from 37 cases and 58 controls using Illumina Human-6 v.2 Expression BeadChip (Illumina, San Diego, CA, USA) according to Illumina's standard protocols. The sample preparation has been described previously,11, 14 and the data set is available at ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae/) (accession code E-TABM-682). To make all transcription values comparable across individuals as well as across genes, these were normalized using rank-based inverse normal transformation as described previously.9
The SAFHS
Study groups
The SAFHS includes 1240 individuals from 42 extended families of Mexican Americans from San Antonio, TX, USA.9, 15, 16 This study aims to quantify the relative contributions of genetic and environmental factors to the risk of developing CVD and participants were not ascertained owing to CVD status. Phenotypic assessment of cardiovascular risk factors has been performed, including serum levels of lipids, lipoproteins, glucose, hormones, adiposity, and blood pressure. Informed consent was obtained from all participants before collection of samples and the study was approved by the University of Texas Health Sciences Center at San Antonio IRB.
Transcriptional profiling and genome-wide SNP data
Transcriptional profiling of lymphocyte-derived RNA has previously been performed on 1240 participants from the SAFHS using the Illumina Sentrix Human Whole Genome (WG-6) Series I BeadChips.9 This array includes 47 289 unique probes, of which 19 648 showed significant expressions in the SAFHS data set. This data set is available at ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae/) (accession code E-TABM-305). In addition, 858 individuals have been genotyped according to the manufacturer's instructions on using the Illumina Human1M-Duo BeadChip (Illumina). Analysis of the raw data was carried out in the BeadStudio software with the recommended parameters for the Infinium assay and using the genotype cluster files provided by Illumina. Samples with a call rate below 97%, SNPs with call rate below 95%, and SNPs deviating from Hardy–Weinberg equilibrium (P<1.0 × 106) were excluded.
Statistical analyses
Differentially expressed genes were identified using the stats library in R. A linear regression model (transcription level ∼PE status+RIN, where RIN is the RNA integrity number) was fitted for each transcript using the lm() function. Summary statistics were computed for the fitted linear model using the summary.lm() function and the P-values were extracted based on t-statistics. False discovery rate (FDR)17 was calculated using the fdrtool() function implemented in the fdrtool R library.18 For downstream analyses, only the differentially expressed genes (FDR P-value <0.05) were included. The correlation between the transcription level and the cardiovascular phenotypes (high-density lipoprotein-C, low-density lipoprotein-C, triglycerides, and total cholesterol) were estimated using a maximum-likelihood test for family data, implemented in SOLAR.19 Cis-regulating SNPs and SNPs associated with the lipid traits (high-density lipoprotein-C, low-density lipoprotein-C, triglycerides, or total cholesterol) were identified using an additive model of association for family data, implemented in SOLAR.19
RESULTS
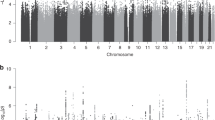
An overview of the study design is presented in Table 1 and the clinical characteristics of the PE study group can be found in Supplementary Table S1. Whole genome expression profiling in decidua basalis tissue from Norwegian preeclamptic cases and controls identified 24 625 transcripts that were detected above background signals (P<0.01), out of the 48 701 probes. In total, 222 genes (Supplementary data Table S2) were differentially expressed between the PE cases and controls (FDR P-value <0.05). For the whole genome profiling in the SAFHS, 19 648 transcripts were detected above the background signals (P<0.01), out of 47 289 unique probes. In total, 109 of the 222 transcripts that were differentially expressed in the decidual tissues were significantly expressed in the lymphocyte data from the SAFHS (Supplementary Table S2).
Correlation of leukocyte gene expression levels with lipid levels
The transcription levels of 10 out of 109 genes tested were associated (FDR P-value <0.05) with one or more of the lipid traits (Table 2). Although the P-value for most transcripts fell just below the threshold of significance, the correlation between expression of ACOX2 (acyl-coenzyme A oxidase 2, branched chain) and triglyceride levels was highly significant (P=5.64 × 10−7; FDR P-value=0.00017). The transcription level of ACOX2 is inversely correlated (negative β) with both triglyceride levels and PE status (Table 2).
ACOX2 is cis-regulated and genetic variation is associated with triglyceride levels
We used the genotype data from the SAFHS to identify proximal cis-regulatory SNPs (SNPs that regulate the transcription level of the gene they are located in). There were eight SNPs on the BeadChip that were within the gene, in 5′- or 3′-untranslated region or close to (10 kb upstream) the ACOX2 gene (Table 3). One SNP (rs4681689) was associated (P=0.0051) with triglyceride levels. Two others (rs1127745 and rs13434020) that are in complete linkage disequilibrium were associated with the transcription level of the gene (P=0.0045). Adjusting for multiple testing (seven independent tests), the associations with triglyceride, and expression levels remained statistically significant after Bonferroni correction (adjusted P=0.032 vs 0.036).
DISCUSSION
We have shown that the ACOX2 gene is downregulated in decidual tissue collected from Norwegian women with PE (negative β; Table 2). We have also found that the transcription level of ACOX2 in leukocytes from SAFHS to be inversely correlated (negative β; Table 2) with triglyceride levels. Increased triglyceride levels are a well-known independent risk factor for CVD,20 and increased triglyceride levels has also been reported in PE.21, 22, 23 We propose that increased triglyceride levels caused by the downregulation of ACOX2 to be a possible shared genetic risk factor for PE and CVD.
Peroxisomes are one of the main locations for lipid metabolism in human beings.24 They are involved in the β-oxidation of very long straight-chain fatty acids, branched-chain fatty acids, dicarboxylic fatty acids, and eicosanoids, as well as in the β-oxidation of the side chain of the bile acid intermediates, resulting in the formation of the primary bile acids. These different substrates are likely to be degraded by distinct oxidation pathways, where the first and rate-limiting step is carried out by two acyl-CoA oxidases.25 However, ACOX2 oxidizes the CoA esters of 2-methyl-branched fatty acids and of the bile acid intermediates di- and trihydroxycoprostanic acids. Insufficient levels of ACOX2 are likely to affect the bile acid production by increasing the fraction of bile acid intermediates to mature bile acids. Furthermore, it has been shown that mature bile acid is an activator of the farnesoid X receptor (F × R) leading to reduced levels of triglycerides,26 which suggests that FXR plays a critical role in lipid metabolism. Thus, our observation of the highly inverse correlation between the transcription levels of ACOX2 and triglyceride levels suggests that ACOX2 may act as a regulator triglyceride levels through the FXR.
Hypertriglyceridemia can elevate free fatty acid concentrations and decrease low-density lipoprotein particle size giving rise to lipid peroxidation, one of the important lipid abnormalities seen in PE.27 Increased lipid peroxidation may confer PE susceptibility by endothelial cell activation.21 Similarly, triglyceride-rich lipoproteins have been suggested to trigger endothelial dysfunction28 and artherothrombosis,29 and they also play an important role in thrombin formation and may induce platelet aggregation, which can contribute to coagulopathy, another characteristic of PE.30 An alternative explanation to downregulation of ACOX2 in PE might be through the bile acid intermediates. Although ACOX2 plays a direct role in the formation of bile acids, it is likely that there also is a connection between ACOX2 and the level of bile acid intermediates. Some bile acid intermediates promote generation of reactive oxygen species leading to oxidative stress.31 A number of studies indicate that oxidative stress plays an important role in the etiology of PE32, 33 and CVD.34
In our study, we identified SNPs associated with transcriptional regulation of the ACOX2 gene and with variation in serum triglyceride levels. Our data suggest that the ACOX2 gene harbors regulatory and potentially functional variants that may be of relevance to the regulation of triglyceride levels and to disease susceptibility. Recently, a large number of genome-wide association studies has been performed and hundreds of SNPs have been identified to be associated with different human traits, including serum triglyceride levels.35 To our knowledge, no association between triglycerides and SNPs in ACOX2 has been seen. However, the fraction of the heritability explained by the SNPs identified by genome-wide association studies is low, and most of the variation behind human traits remains unknown.35 This might be due to rare variants, which are under-represented in genome-wide association studies, playing an important role in phenotypic variation, or due to incomplete linkage disequilibrium between the typed markers and the causal variants. To date, more than 200 SNPs have already been identified in the ACOX2 gene region and the linkage disequilibrium in the region is modest. Consequently, the true regulatory and functional SNPs within the region are not likely to be present within our set of eight SNPs (which is typically typed in most genome-wide association studies), and resequencing and/or additional SNP typing is required to be able to identify these variants. In our study, both phenotypes (PE and triglyceride levels) are correlated with expression levels to a much higher extent than to the available SNPs. Consequently, it is tempting to speculate that the regulation of ACOX2 expression is not dramatically influenced by any one SNP, but rather multiple factors, for example, epigenetic and trans-regulation play contributory roles. It has also been shown previously that the methylation pattern of many genes differ between placentas from women with PE compared to controls.36
In addition to ACOX2, we identified nine other genes whose transcription levels were correlated to both PE status and lipid levels (Table 2). Even though these show lower statistical significance compared with ACOX2, we cannot exclude any of these genes from being as important in relation to developing both PE and CVD. Two of these genes in particular are worth noting. NOD1 and SPPL2A both play an important role in the immune response and the development of inflammatory disease37, 38 and are therefore plausible candidate risk genes for PE and CVD.
One limitation to our study is that the PE cases and controls are not matched with regard to gestational age (Supplementary Table S1). It has been reported that the expression level changes dramatically for some genes between mid-gestation and term.39 However, among our top genes (Table 1) only one, KIAA1598, has previously been shown to differ between term and mid-gestation,39 and there is no indication that expression of ACOX2 should be influenced by gestational age. In addition, women giving birth pre-term without any indications of PE are likely to suffer from some other health complication (eg, infections)40 that might as well influence the transcription level. There are also other factors that do differ slightly (not significant) between groups (Supplementary Table S1). However, including additional covariates, such as body mass index, maternal and infant sex and parity in the regression analyses did not change the results (data not shown). We have not performed validation of the expression values in our study, primarily due to lack of RNA available for these validation experiments. However, in other studies, microarrays have been shown to give accurate detection of transcripts with a high correlation to reverse transcription-PCR verifications.11 The normalization procedure for reverse transcription-PCR uses housekeeping genes whose expression in fact varies dramatically between individuals.9 Consequently, the normalization carried out for the microarray experiment, based on the average expression level per individual, is likely to be much more accurate. Genetic and environmental factors might differ between Norway and Texas/Mexico. Also, the major difference between the samples is the different tissues studied (basalis tissues vs blood lymphocytes) as the expression of some genes is tissue specific. This also explains that 109 of 222 PE transcripts were expressed above the background level in the lymphocytes.
In summary, we have identified ACOX2 to be downregulated in decidual tissues from women with PE and the expression level of the gene in peripheral lymphocytes to be inversely correlated with circulating triglyceride levels. In addition, SNPs within the ACOX2 gene appears to influence both the expression of the gene as well as serum triglyceride levels. The involvement of ACOX2 in bile acid formation, and subsequent association of bile acids and their intermediates with both oxidative stress and regulation of triglyceride levels makes ACOX2 a likely candidate gene in the pathogenesis of both PE and CVD.
Accession codes
References
Roberts JM, Pearson G, Cutler J, Lindheimer M : Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension 2003; 41: 437–445.
Goldenberg RL, Rouse DJ : Prevention of premature birth. N Engl J Med 1998; 339: 313–320.
Craici I, Wagner S, Garovic VD : Preeclampsia and future cardiovascular risk: formal risk factor or failed stress test? Ther Adv Cardiovasc Dis 2008; 2: 249–259.
Magnussen EB, Vatten LJ, Lund-Nilsen TI, Salvesen KA, Davey Smith G, Romundstad PR : Prepregnancy cardiovascular risk factors as predictors of pre-eclampsia: population based cohort study. BMJ 2007; 335: 978.
Sattar N, Bendomir A, Berry C, Shepherd J, Greer IA, Packard CJ : Lipoprotein subfraction concentrations in preeclampsia: pathogenic parallels to atherosclerosis. Obstet Gynecol 1997; 89: 403–408.
Irgens HU, Reisaeter L, Irgens LM, Lie RT : Long term mortality of mothers and fathers after pre-eclampsia: population based cohort study. BMJ 2001; 323: 1213–1217.
Funai EF, Friedlander Y, Paltiel O et al: Long-term mortality after preeclampsia. Epidemiology 2005; 16: 206–215.
Salonen Ros H, Lichtenstein P, Lipworth L, Cnattingius S : Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am J Med Genet 2000; 91: 256–260.
Goring HH, Curran JE, Johnson MP et al: Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet 2007; 39: 1208–1216.
Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS, Williams MA : Differential placental gene expression in preeclampsia. Am J Obstet Gynecol 2008; 199: 566–568.
Løset M, Mundal SB, Johnson MP et al: A transcriptional profile of the decidua in preeclampsia. Am J Obstet Gynecol 2011; 204: 84–86.
Winn VD, Gormley M, Paquet AC et al: Severe preeclampsia-related changes in gene expression at the maternal-fetal interface include sialic acid-binding immunoglobulin-like lectin-6 and pappalysin-2. Endocrinology 2009; 150: 452–462.
Gifford RW, August PA, Cunningham G et al: Report of the National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 2000; 183: S1–S22.
Fenstad MH, Johnson MP, Loset M et al: STOX2 but not STOX1 is differentially expressed in decidua from preeclamptic women. Mol Hum Reprod 2010; 16: 960–968.
MacCluer JW, Stern MP, Almasy L et al: Genetics of atherosclerosis risk factors in Mexican Americans. Nutr Rev 1999; 57: S59–S65.
Mitchell BD, Kammerer CM, Blangero J et al: Genetic and environmental contributions to cardiovascular risk factors in Mexican Americans. The San Antonio Family Heart Study. Circulation 1996; 94: 2159–2170.
Hochberg Y, Benjamini Y : More powerful procedures for multiple significance testing. Stat Med 1990; 9: 811–818.
Strimmer K : A unified approach to false discovery rate estimation. BMC Bioinform 2008; 9: 303.
Almasy L, Blangero J : Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 1998; 62: 1198–1211.
Criqui MH, Heiss G, Cohn R et al: Plasma triglyceride level and mortality from coronary heart disease. N Engl J Med 1993; 328: 1220–1225.
Chappell LC, Seed PT, Briley A et al: A longitudinal study of biochemical variables in women at risk of preeclampsia. Am J Obstet Gynecol 2002; 187: 127–136.
Gratacos E, Casals E, Sanllehy C, Cararach V, Alonso PL, Fortuny A : Variation in lipid levels during pregnancy in women with different types of hypertension. Acta Obstet Gynecol Scand 1996; 75: 896–901.
Hubel CA, McLaughlin MK, Evans RW, Hauth BA, Sims CJ, Roberts JM : Fasting serum triglycerides, free fatty acids, and malondialdehyde are increased in preeclampsia, are positively correlated, and decrease within 48 h post partum. Am J Obstet Gynecol 1996; 174: 975–982.
Reddy JK, Mannaerts GP : Peroxisomal lipid metabolism. Annu Rev Nutr 1994; 14: 343–370.
Casteels M, Schepers L, Van Veldhoven PP, Eyssen HJ, Mannaerts GP : Separate peroxisomal oxidases for fatty acyl-CoAs and trihydroxycoprostanoyl-CoA in human liver. J Lipid Res 1990; 31: 1865–1872.
Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B : Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009; 89: 147–191.
Hubel CA, Roberts JM, Taylor RN, Musci TJ, Rogers GM, McLaughlin MK : Lipid peroxidation in pregnancy: new perspectives on preeclampsia. Am J Obstet Gynecol 1989; 161: 1025–1034.
Kugiyama K, Doi H, Motoyama T et al: Association of remnant lipoprotein levels with impairment of endothelium-dependent vasomotor function in human coronary arteries. Circulation 1998; 97: 2519–2526.
Mochizuki M, Takada Y, Urano T et al: The in vitro effects of chylomicron remnant and very low density lipoprotein remnant on platelet aggregation in blood obtained from healthy persons. Thromb Res 1996; 81: 583–593.
Wetzka B, Winkler K, Kinner M, Friedrich I, Marz W, Zahradnik HP : Altered lipid metabolism in preeclampsia and HELLP syndrome: links to enhanced platelet reactivity and fetal growth. Semin Thromb Hemost 1999; 25: 455–462.
Ferdinandusse S, Denis S, Dacremont G, Wanders RJ : Toxicity of peroxisomal C27-bile acid intermediates. Mol Genet Metab 2009; 96: 121–128.
Takagi Y, Nikaido T, Toki T et al: Levels of oxidative stress and redox-related molecules in the placenta in preeclampsia and fetal growth restriction. Virchows Arch 2004; 444: 49–55.
Tranquilli AL, Bezzeccheri V, Giannubilo SR, Scagnoli C, Mazzanti L, Garzetti GG : Amniotic vascular endothelial growth factor (VEGF) and nitric oxide (NO) in women with subsequent preeclampsia. Eur J Obstet Gynecol Reprod Biol 2004; 113: 17–20.
Madamanchi NR, Vendrov A, Runge MS : Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 2005; 25: 29–38.
Teslovich TM, Musunuru K, Smith AV et al: Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713.
Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson WP : DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet 2010; 18: 1006–1012.
Friedmann E, Hauben E, Maylandt K et al: SPPL2a and SPPL2b promote intramembrane proteolysis of TNFalpha in activated dendritic cells to trigger IL-12 production. Nat Cell Biol 2006; 8: 843–848.
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE : Nod-like proteins in immunity, inflammation and disease. Nat Immunol 2006; 7: 1250–1257.
Winn VD, Haimov-Kochman R, Paquet AC et al: Gene expression profiling of the human maternal–fetal interface reveals dramatic changes between midgestation and term. Endocrinology 2007; 148: 1059–1079.
Romero R, Espinoza J, Kusanovic JP et al: The preterm parturition syndrome. BJOG 2006; 113 (Suppl 3): 17–42.
Acknowledgements
This work was supported by grants from FUGE – Functional genomics in Norway within the Research Council of Norway (ÅJ, MHF), by motility grant from NTNU – The Norwegian University of Science and Technology (ÅJ), Sven and Dagmar Saléns foundation (ÅJ) and the Fulbright foundation for educational exchange (MHF). The work was also supported by National Institutes of Health grants (grant number HD049847) (to EKM, SPB). This investigation was conducted in facilities constructed with support from Research Facilities Improvement Program grants (grant numbers RR13556, RR017515) from the National Center for Research Resources, National Institutes of Health. The SAFHS is supported by NLBI (grant number HL45522) and SOLAR software is supported by NIMH (grant number MH59490). We would like to thank Mari Löset, Siv B Mundal and everyone who have contributed to produce the data that are used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Johansson, Å., Curran, J., Johnson, M. et al. Identification of ACOX2 as a shared genetic risk factor for preeclampsia and cardiovascular disease. Eur J Hum Genet 19, 796–800 (2011). https://doi.org/10.1038/ejhg.2011.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.19
Keywords
This article is cited by
-
Ancestry-related distribution of Runs of homozygosity and functional variants in Qatari population
BMC Genomic Data (2022)
-
Cardiovascular Dysfunction in Intrauterine Growth Restriction
Current Hypertension Reports (2022)
-
Preeclampsia and Related Cardiovascular Risk: Common Genetic Background
Current Hypertension Reports (2018)
-
The Impact of Maternal-Fetal Genetic Conflict Situations on the Pathogenesis of Preeclampsia
Biochemical Genetics (2015)
-
Race-associated biological differences among Luminal A breast tumors
Breast Cancer Research and Treatment (2015)
