
Abstract
Pontocerebellar hypoplasia (PCH) is a group of autosomal recessive neurodegenerative disorders characterized by prenatal onset of stunted brain growth and progressive atrophy predominantly affecting cerebellum, pons and olivary nuclei, and to a lesser extent also the cerebral cortex. Six subtypes (PCH1–6) were described and genes for four types (PCH1, 2, 4 and 6) were identified. Mutations in the tRNA splicing endonuclease subunit (TSEN) genes 54, 2 and 34 are found in PCH2 and PCH4. One family with severe prenatal onset of PCH has been the only representative of PCH5 published so far, and the molecular genetic status of PCH5 has not been ascertained until now. We screened the previously reported PCH5 family for mutations in the TSEN54 gene. The PCH5 patient was found to be the result of compound heterozygosity for the common TSEN54 mutation (p.A307S) plus a novel splice site mutation. The mutations associated with PCH5 are similar to what has been reported in PCH4. Thus, PCH5, PCH4 and PCH2 represent a spectrum of clinical manifestations caused by different mutations in the TSEN genes. We, therefore, propose to classify PCH2, PCH4 and PCH5 as TSEN mutation spectrum disorders.
Similar content being viewed by others

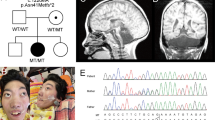
Introduction
Pontocerebellar hypoplasias (PCHs) are a group of lethal autosomal recessive disorders characterized by prenatal onset of stunted growth and atrophy of the cerebellum, pons and olivary nuclei. Six subtypes have been identified so far, each with a distinct pathology and clinical presentation. Microcephaly and delayed development are features of all subtypes. Life expectancy is difficult to predict as survival ranges from early death (1 day) to adult death.1 Mutations in different genes have been identified in four of them: PCH1, PCH2, PCH4 and PCH6.1, 2, 3, 4
PCH2 is the most common subtype and is characterized by jitteriness and the development of dyskinesia and choreatic movements. Other criteria include swallowing problems, central visual failure and the absence of primary optic atrophy. Patients usually die during childhood. Mutations in genes encoding the tRNA splicing endonuclease complex (TSEN) are responsible for PCH2. In most cases an alanine to serine substitution (p.A307S) in TSEN54 is found, whereas rare cases show mutations in TSEN2 and TSEN34.1, 3, 5
PCH4 presents a more severe form of PCH including hypertonia and severe clonus and usually more pronounced cerebellar hypoplasia (see Table 1). Nine cases of genetically confirmed PCH4 have been described so far, of which eight were compound heterozygote for a nonsense mutation or a splice site mutation and the common p.A307S missense mutation in the TSEN54 gene.1 One case carried a rare missense mutations for TSEN54 on one allele (p.S93P) in addition to homozygosity for the common p.A307S mutation. Thus, a reduced amount of, or aberrant, TSEN54 protein is present in PCH4 patients, resulting in a more severe phenotype in comparison with PCH2. In seven of eight PCH4 cases, the Magnetic Resonance Imaging (MRI) scans showed immaturity of the cerebral cortex with underdeveloped cerebral hemispheres and increased extracerebral cerebrospinal fluid volume. The cerebellar pathology in PCH4 is also more severe compared with PCH2. In addition to the severely affected cerebellar hemispheres, six out of eight cases showed mild or severe atrophy of the cerebellar vermis.1 In 2006, Patel et al6 presented a family with a new PCH type identified as PCH5, with fetal seizures and on autopsy marked degeneration of the cerebellar vermis. The parents were healthy and non-consanguineous, with three children affected with severe olivopontocerebellar hypoplasia, two healthy siblings and a miscarriage at 12 weeks gestation. The pedigree was consistent with an autosomal recessive mode of inheritance. All affected cases suffered from intrauterine seizure-like movements, recognized by the mother as rhythmic movements and confirmed on fetal ultrasound. Patient 1 died from apnea at the age of 3 days after withdrawal of care. She suffered from generalized hypertonia, sustained clonus and respiratory distress. The pregnancies with patients 2 and 3 were terminated at 27 and 20 weeks, respectively, following the finding of clearly visible cerebellar hypoplasia on ultrasound. Autopsies done on these three affected cases revealed immature neurons in the pons, with gliosis and apoptosis in patients 1 and 2 and a C-shaped inferior olive present in all cases.6 The cerebellar vermis and hemispheres were severely reduced. The vermis showed absence and immaturity of Purkinje cells and distinct focal absence of the external granule cell layer. Based on the clinical and pathological manifestations, Patel et al6 proposed that this family constitute a new subtype of PCH, PCH5.
Methods
To test whether PCH5 is part of the TSEN mutation spectrum, we tested patient 1 from the report of Patel et al6 for TSEN54 mutations with parental consent. PCR products were sequenced using the Big Dye Terminator Sequencing kit and ABI PRISM 3730XL DNA Analyser (Applied Biosystems, Foster City, CA, USA). Codon Code Software version 3.5.6 (CodonCode Corporation, Dedham, MA, USA) was used to analyse the sequenced samples.
Results and discussion
Analysis of patient 1 and his parents showed that the index case was a compound heterozygote for two TSEN54 mutations. One allele carried the common PCH-associated mutation c.919G>T, p.A307S and the other a splice site mutation; c.468+2T>C. This variant was not found in 176 control chromosomes. Homozygosity for the p.A307S TSEN54 mutation results in PCH2.3 The c.468+2T>C is located in the donor splice site of intron 5, which makes skipping of exon 5 likely. This was confirmed with Alamut software, this tool uses four different splice site prediction algorithms (http://www.interactive-biosoftware.com/alamut.html). The character of these mutations is similar to those reported in PCH4. Comparing the clinical and pathological features of PCH5 with a genetically confirmed group of PCH4 patients showed that the findings are similar.1 For example, the inferior olivary nuclei in the PCH5 family reported by Patel et al6 showed a C-shaped structure similar to what is reported in PCH4.7, 8, 9 The absence of the external granular layer in the vermis and cerebellar hemispheres described in PCH5 is focal and similar to the segmental loss of cerebellar cortex typically seen in PCH2 and PCH4.8 In the PCH5 cases, the vermis was noted to be more affected than the hemispheres; however, as the vermal cortex is only relatively spared in PCH4 cases there is no essential difference. In our recent study, six out of eight PCH4 cases, on whom brain MRI was available, also showed mild or severe atrophy of the cerebellar vermal folia.1 The prenatal seizure-like activity observed in the PCH5 cases is similar to the severe neonatal clonus seen in PCH4.9 Although milder, the clinical findings in PCH2 are similar to what is reported in PCH4 and PCH5.10 Early postnatal lethality due to hypoventilation, as observed in the firstborn of the PCH5 family, is also commonly observed in PCH4.1
Thus, our molecular genetic findings and the phenotype in PCH5 are similar to that of PCH4. As PCH2 is also caused by mutations in one of the TSEN subunits, we propose that PCH2, PCH4 and PCH5 will be united as a spectrum of clinical manifestations caused by different mutations in the TSEN genes (Table 1). We, therefore, propose to reassign PCH2, PCH4 and PCH5 as TSENopathies.
References
Namavar Y, Barth PG, Kasher PR et al: Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 2011; 134: 143–156.
Edvardson S, Shaag A, Kolesnikova O et al: Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007; 81: 857–862.
Budde BS, Namavar Y, Barth PG et al: tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 2008; 40: 1113–1118.
Renbaum P, Kellerman E, Jaron R et al: Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 2009; 85: 281–289.
Cassandrini D, Biancheri R, Tessa A et al: Pontocerebellar hypoplasia: clinical, pathologic, and genetic studies. Neurology 2010; 75: 1459–1464.
Patel MS, Becker LE, Toi A, Armstrong DL, Chitayat D : Severe, fetal-onset form of olivopontocerebellar hypoplasia in three sibs: PCH type 5? Am J Med Genet A 2006; 140: 594–603.
Albrecht S, Schneider MC, Belmont J, Armstrong DL : Fatal infantile encephalopathy with olivopontocerebellar hypoplasia and micrencephaly. Report of three siblings. Acta Neuropathol 1993; 85: 394–399.
Barth PG, Aronica E, de Vries L et al: Pontocerebellar hypoplasia type 2: a neuropathological update. Acta Neuropathol 2007; 114: 373–386.
Chaves-Vischer V, Pizzolato GP, Hanquinet S, Maret A, Bottani A, Haenggeli CA : Early fatal pontocerebellar hypoplasia in premature twin sisters. Eur J Paediatr Neurol 2000; 4: 171–176.
Barth PG, Blennow G, Lenard HG et al: The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly, and extrapyramidal dyskinesia (pontocerebellar hypoplasia type 2): compiled data from 10 pedigrees. Neurology 1995; 45: 311–317.
Steinlin M, Klein A, Haas-Lude K et al: Pontocerebellar hypoplasia type 2: variability in clinical and imaging findings. Eur J Paediatr Neurol 2007; 11:146–152.
Acknowledgements
Financial support was kindly provided by the Hersenstichting Nederland KS2009(1)-81.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Namavar, Y., Chitayat, D., Barth, P. et al. TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur J Hum Genet 19, 724–726 (2011). https://doi.org/10.1038/ejhg.2011.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.8
Keywords
This article is cited by
-
TSEN54 Gene-Related Pontocerebellar-Hypoplasia and Role of Prenatal MR Imaging: Besides the Common Posterior Fossa Cystic Malformations
The Cerebellum (2022)
-
Diagnostic Approach to Cerebellar Hypoplasia
The Cerebellum (2021)
-
Pontocerebellar Hypoplasia: a Pattern Recognition Approach
The Cerebellum (2020)
-
What’s new in pontocerebellar hypoplasia? An update on genes and subtypes
Orphanet Journal of Rare Diseases (2018)
-
Update on neuroimaging phenotypes of mid-hindbrain malformations
Neuroradiology (2015)
