
Abstract
Fragile X syndrome is the most common inherited form of mental retardation. It is caused by expansion of a trinucleotide (CGG)n repeat sequence in the 5′ untranslated region of the FMR1 gene, resulting in promoter hypermethylation and suppression of FMR1 transcription. Additionally, pre-mutation alleles in carrier males and females may result in Fragile X tremor ataxia syndrome and primary ovarian insufficiency, respectively. Fragile X is one of the most commonly requested molecular genetic tests worldwide. Quality assessment schemes have identified a wide disparity in allele sizing between laboratories. It is therefore important that clinical laboratories have access to characterized reference materials (RMs) to aid accurate allele sizing and diagnosis. With this in mind, a panel of genotyping RMs for Fragile X syndrome has been developed, which should be stable over many years and available to all diagnostic laboratories. Immortalized cell lines were produced by Epstein–Barr virus transformation of lymphocytes from consenting patients. Genomic DNA was extracted in bulk and RM aliquots were freeze-dried in glass ampoules. Twenty-one laboratories from seventeen countries participated in a collaborative study to assess their suitability. Participants evaluated the samples (blinded, in triplicate) in their routine methods alongside in-house and commercial controls. The panel of five genomic DNA samples was endorsed by the European Society of Human Genetics and approved as an International Standard by the Expert Committee on Biological Standardization at the World Health Organization.
Similar content being viewed by others

Introduction
Fragile X syndrome is the most common heritable cause of mental retardation, affecting approximately 1 in 5000 males and 1 in 10 000 females. It is caused by the expansion of a tract of CGG repeats in the 5′-untranslated region of the Fragile X mental retardation 1 (FMR1) gene located at Xq27.3, with consequent hypermethylation of promotor regions and silencing of gene expression.1 As the phenotypic presentation and frequency of clinical signs are variable, clinical diagnosis is challenging, and definitive diagnosis in suspected individuals requires molecular measurement of the (CGG)n allele size. The size of the (CGG)n repeat tract is highly polymorphic in the general population, containing from 6 to about 50 repeats and most commonly 29–30 repeats.2, 3, 4 Individuals with alleles of 59–200 repeats, known as pre-mutations, are considered to be carriers, as alleles of this length are highly unstable in maternal transmission and are at very high risk of expansion into ‘full mutation’ alleles of >200 repeats, which are causative of the Fragile X syndrome.
Selected screening is highly justified among mentally retarded individuals, not only to provide the benefits of early clinical intervention but also to enable prenatal diagnosis to be offered to related carriers.5, 6 Genetic testing for Fragile X syndrome is widespread, but it has been acknowledged by leading laboratories in the field to be one of the most technically challenging genetic tests. Performance in proficiency testing has been shown to be sub-optimal in several EQA schemes.7, 8 Any assay using PCR amplification to determine (CGG)n must overcome the problem of amplifying long sections of repetitive CG-rich sequence. Validation of the assays used is difficult because of the lack of reliable reference materials (RMs). Accurate allele sizing becomes essential when distinguishing between alleles around the normal/pre-mutation and pre-mutation/full mutation thresholds. Most laboratories use DNA samples from patients who have been characterized by their own laboratory as controls. As such, allele sizing accuracy may vary between laboratories. Laboratories that are new to the field often rely on small finite amounts of materials supplied by other genetic reference laboratories, for example as part of external quality assessment schemes. However, these control materials are in short supply because of the relatively large amount needed for investigation (by Southern blotting) and sample distribution restrictions in Europe imposed by the IVD Directive (Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices).
The high frequency of male and female carriers of FMR1 pre-mutation alleles (∼1/813–1/251 and ∼1/259–1/113, respectively,)9, 10, 11, 12, 13 and the occurrence of pre-mutation phenotypes such as Fragile X tremor ataxia syndrome (FXTAS)14 and Fragile X-associated primary ovarian insufficiency (FXPOI)15 will doubtless lead to increased FMR1 genotyping and possibly to screening of the general population. The case for certified RMs to aid accuracy in FMR1 genotyping is therefore increasing in strength.
There are no internationally certified genetic RMs available for in vitro diagnosis of Fragile X syndrome. A panel of nine PCR amplicons with a variety of FMR1 trinucleotide repeat length standards is available from the US National Institute of Standards and Technology (NIST). However, the panel does not contain any full mutations and the materials can only be used in PCR and not in the more definitive Southern blot assay. Genomic DNA (gDNA) RMs would be preferable as these more closely resemble patient specimens. Well-characterized genomic DNA materials are available from Coriell Cell Repositories but are not approved or intended for in vitro diagnostic use.16 Furthermore, there may be differences in DNA quality and performance (and also possibly even differences in allele sizes due to somatic cell mutation17 (also, KEW and JRH, unpublished data)) in future batches.
We have therefore chosen to produce in bulk a panel of gDNA materials that could be certified and, when eventually required, can be replenished from the same source. Bulk genomic DNA was extracted from immortalized cell lines produced by Epstein–Barr virus (EBV) transformation of lymphocytes from donors who were known to carry the wild type, pre-mutation and full mutation genotypes. Following validation by an international collaborative study, a panel of five gDNAs was endorsed by the European Society of Human Genetics and then approved as an International Standard by the Expert Committee on Biological Standardization at the World Health Organization (WHO) in November 2008.
Materials and methods
Patients and cell lines
A protocol for approaching patients, obtaining informed consent and anonymizing samples was approved by a local research ethics committee. Patient blood samples were collected from six consenting donors as part of ‘CRMGEN’, an EU 5th framework project to evaluate and develop certified RMs for genetic testing. One donor was a normal female individual and the remainder had been identified after previous molecular genetic investigation. Classifications of these patients were: female pre-mutation, male pre-mutation, male full mutation and female full mutation ( × 2). Lymphoblastoid cell lines were established after EBV transformation, ensuring continued future supply. Each cell line was tested for Hepatitis A, Hepatitis B, Hepatitis C, Parvovirus B19 and HIV1. Master and working cell banks were established and stored to ensure continual future supplies of the same cells. Safety of storage was guaranteed by distribution to multiple liquid nitrogen vessels at multiple locations. Cell banks were identity-tested by DNA profiling with the Applied Biosystems 11-locus SGM+ kit. FMR1 (CCG)n repeat genotypes of the cell lines were confirmed to match those of the donor patients, except for the two female full mutation cell lines that remained unconfirmed before the collaborative study. For that reason, two female full mutation samples were included in the panel for the collaborative study, in case one proved more suitable than the other for use as an RM.
DNA extraction and freeze drying
Cells were bulk cultured (ECACC, Porton Down, UK) to approximately 1010 cells and frozen as pellets of 108 cells. Genomic DNA was extracted from cell pellets using the PureGene DNA purification and Autopure LS equipment (Qiagen, Crawley, UK). The integrity of the extracted DNA was confirmed by electrophoresis in agarose gels. Each of the DNA samples was dissolved at a concentration of approximately 29 μg/ml in 0.8 mM Tris/0.08 mM EDTA buffer with 2.5 mg/ml Trehalose. A volume of 0.8 ml of this solution was dispensed into a minimum of 1200 glass ampoules and freeze-dried. Sealed ampoules were stored at −20 °C.
Homogeneity testing
Samples were tested for homogeneity by determining DNA concentration by spectrophotometric absorbance analysis (Nanodrop, Wilmington, DE, USA). After reconstitution in distilled water, the samples were allowed to equilibrate to room temperature and mixed through pipetting after 30 min. Samples were quantitated after 1 h. The accuracy and precision of the spectrophotometer were determined by performing multiple reads on a control sample of commercial genomic DNA (Promega, Southampton, UK).
Accelerated degradation study
Ampoules were tested for degradation by agarose gel and quantitative PCR comparison of samples stored at +56 and −20 °C with samples stored at −150 °C. For agarose gel electrophoresis, the size marker was Hyper Ladder VI (Bioline, London, UK). For Q-PCR, the PCR primer sequences (5′–3′), forward: 5′-TTACAAGCCTGATGAAGGGA-3′ and reverse: 5′-CCATGAATAGCACTGGGAGCATTGAAGC-3′, were used at 10 μ M to amplify a 486-bp sequence of F2 on chromosome 11. Cycling conditions were 95 °C for 10 min, then 45 cycles of 95 °C for 10 s, 60 °C for 20 s and 72 °C for 30 s using the LightCycler FastStart DNA Master plus SYBR Green I kit on a LightCycler 480 thermal cycler (Roche, Burgess Hill, UK).
Methylation analysis
For analysis of methylation status at the FMR1 promoter, Methyl Primer Express v1.0 was used to design bisulphite-specific sequence primers for the amplification of a 192-bp region, covering 22 CpGs, of the FMR1 gene promoter region (position 3435241–3437401 of NT_011681 of Chromosome X, NC_000023), upstream of the CGG repeat region. Primers used were forward 5′-TTACAAGCCTGATGAAGGGA-3′ and reverse 5′-CCATGAATAGCACTGGGAGCATTGAAGC-3′. Ampoules were reconstituted in 40 μl water and left to reconstitute for 60 min. Approximately 50 ng of bisulphite-converted DNA was used in the final HRM reaction mix, containing 1 U Hot Star Taq (Qiagen), 3 mM MgCl2, 5 pmol primers, 200 μ M dNTPs and 5 μ M Syto9. PCR was performed on the Rotorgene 6000, and consisted of an initial 10 min denaturation step at 95 °C, followed by 40 cycles of 30 s at 95 °C, 30 s at 55 °C and 45 s at 72 °C with a final 7 min extension at 72°C. This PCR was followed by a melting step for HRM, which consisted of 95 °C for 5 s, followed by 50 °C for 5 min and a temperature ramping from 70 to 95 °C at a rate of 0.2 °C/s. Fluorescence was acquired on the HRM channel. HRM data were analysed using Rotorgene 6000 software. The leading and trailing edges were normalized at 72–74 and 83–86 °C, respectively. The assay was performed on three ampoules of each sample.
Collaborative validation study
Each of the six gDNA materials was sent in triplicate and blinded to each laboratory with instructions for reconstitution and storage. Each laboratory was asked to perform their routine test(s) for Fragile X syndrome on each of the 18 coded samples and to genotype the samples in groups of six on three separate days as indicated on the results sheet, using different lots of reagents or different operators if possible. Laboratories were asked to report the number of trinucleotide repeats. The overall findings for each sample and raw data, eg-images of autoradiographs, were to be returned together with full details of techniques used, any in-house or commercial controls and reasons for failure of any of the samples tested.
Results
Each cell line was found to be negative for Hepatitis A, Hepatitis B, Hepatitis C, Parvovirus B19 and HIV1. All EBV-transformed cell lines remain positive for the presence of the EBV genome. Whereas there is an unquantified risk of EBV infectivity in the cell lines, infective viral particles are unlikely to survive DNA extraction and have not been detected in electron microscopy studies or in infectivity assays (JRH, unpublished data). In all cases cell line DNA profiles matched the profile of the original blood samples. No samples showed evidence of cross-contamination. However, sample 07/174 showed a very small additional peak at just two short tandem repeat marker loci. These peaks were not detected in the original blood samples.
Bulk genomic DNA was filled into glass ampoules, freeze-dried and sealed. Upon reconstitution with 40 μl sterile water the DNA concentration was approximately 580 μg/ml in 16 mM Tris, 1.6 mM EDTA and 50 mg/ml Trehalose. The pH of the reconstituted candidate materials was measured and was found to be within 7.4 and 7.8 in all six. Table 1 shows the product summary for the six materials. Residual moisture and oxygen levels were consistent with those obtained in previous fills and within the levels acceptable for International Standards by the WHO.18
Homogeneity
Random ampoules were weighed periodically during the ampoule filling of each material. The coefficient of variation for each fill (0.15–0.31%; Table 1) indicates a very low level of variation in volumes aliquotted during each fill run.
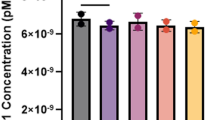
The materials were tested for DNA homogeneity after reconstitution both within ampoules and between ampoules by DNA quantitation. Ten randomly selected ampoules of each material were reconstituted in 40 μl distilled water. The DNA concentration of each ampoule was measured in triplicate. The triplicate reads were highly consistent, giving a modal coefficient of variability of just 2.9%. The distribution of variability is shown in Figure 1a. These data indicate that the material in the ampoules is highly homogeneous 1 h after reconstitution.

Homogeneity. (a) Variability (% CV) in triplicate DNA quantitation reads from 10 ampoules of each material. The modal CV of reads from a single ampoule is 2.9%, indicating the material is highly homogeneous within ampoules. (b) Variability between fills and between ampoules. Columns show mean DNA concentration (μg/ml) from 10 ampoules from each fill. Error bars show DNA concentration variability (±1 SD) between ampoules.
DNA concentrations showed more variation between individual ampoules of each material and between the different materials (Figure 1). However, the intended purpose is independent of DNA concentration above a minimum level and the amount supplied in each ampoule is enough for several tests by Southern blotting. Mean concentrations of each material varied from 568 to 818 μg/ml, equating to a mean of at least 23 μg per ampoule. Coefficients of variability between ampoules of a single material ranged from 7.2 to 15.3%.
Stability
An accelerated degradation study on samples stored at +56 °C was carried out. When compared with samples stored at −150 °C, after 2-years storage there was no detectable degradation in samples 07/122, 07/168, 07/170 or 07/174 as assessed by Q-PCR (Figure 2a). Ampoule 07/120 showed an increase in Cp value of 0.54 between the −150 and +56 °C samples, indicating a 31% decrease in DNA yield. This difference, however, may be the result of ampoule-to-ampoule variability or due to a ‘baking’ effect resulting in incomplete solubilization. The Q-PCR findings were confirmed by agarose gel electrophoresis, where there was a slight increase in band mobility for the +56 °C sample as well as a slight decrease in band intensity, but no visible increase in low molecular weight DNA (Figure 2b). Densitometry analysis of the gel bands indicates an average signal reduction of 22% for the +56 °C samples. Together, the Q-PCR and electrophoresis data indicate some, but limited degradation at +56 °C. However, the limited degradation at +56 °C and the absence of degradation at the storage temperature of −20 °C after 2 years indicate that the DNA should be stable for many years.

Accelerated degradation. (a) Accelerated degradation – real-time PCR. Crossing point (Cp) values for quantitative PCR (Roche Lightcycler) after storage of ampoules at baseline (−150 °C) and elevated temperatures for 2 years. CVs show the level of variability between triplicate runs. (b) Accelerated degradation – agarose gel. 0.7% agarose gel of 200 ng of each sample. The size marker is Hyperladder VI (Bioline).
Validation
To validate that the materials are fit for purpose as RMs in the laboratory diagnosis of Fragile X syndrome, laboratories were invited to participate in an international collaborative study. In all, 38 laboratories were invited to take part in the study, 23 laboratories agreed to participate and results were returned by 21 laboratories. Seventeen different countries were represented among participants who returned the results: 13 from Europe, 4 from North America, 3 from Australasia and 1 from Asia. Collaborative validation study participants were requested to test the 18 coded samples on three separate days using different lots of reagents or different operators if possible. The methods used by the participants are listed in Table 2. Seventeen laboratories used in-house assays and four laboratories used a commercial kit developed by Celera and available from Abbott Molecular (Des Plaines, IL, USA). Some laboratories usually refer selected samples to another laboratory to complete their investigation (laboratories 11 and 15) and others were unable to carry out their usual testing protocol because of technical difficulties (laboratories 8, 14 and 19). Laboratory 14 could not obtain any useful Southern blotting results with the samples supplied, whereas their in-house controls gave the expected results, as did the study samples in their PCR test.
No two in-house methods were entirely identical. Some laboratories indicated that their method was based on a previous publication (see Supplementary Data). Eight laboratories were able to use more than one batch of reagents and the testing was carried out by more than one operator in five laboratories. There is no evidence of the study materials performing differently between different methods. The materials performed similarly to patient samples in Southern blotting, with a spread of bands from expanded alleles, although none of the spreads were as extreme as can be seen in some patient samples (Figure 3a). The methylation states of the alleles are indicated in Figure 3b. The normal female and female pre-mutation samples gave a two-peak melting profile, with the larger peak indicating unmethylated and the smaller peak indicating methylated. This melting profile corresponds with the two alleles present in these samples, one of which is X-inactivated and appears as the higher Tm methylated peak. The female full mutation-1 sample also gave the two-peak melting profile, which indicates that the FMR1 expansion lies largely on the inactivated X-chromosome. In contrast, the female full mutation-2 sample gave a single methylated melting peak profile, indicating that the FMR1 expansion lies largely on the active X-chromosome. Male samples, however, have a simpler melting profile as they possess only a single X allele. The male pre-mutation sample gave a single low Tm peak, indicating that the Fragile X promoter is unmethylated. The male full mutation sample shows a single fully methylated peak, indicating the presence of only a fully methylated Fragile X promoter.

Southern blot of study samples. (a) Southern blot of study samples showing male and female; normal, pre-mutation and full expansion banding patterns. The control samples are patient DNA samples extracted from blood. Consensus expanded triplet repeat numbers are indicated on the right. (b) High-resolution melt analysis. The x-axis represents temperature from 70 to 95 °C. The y-axis represents dF/dT (difference in fluorescence over temperature) from zero to 1.4. Methylation status is indicated by the presence and relative size of the left peak (unmethylated) and the right peak (methylated). The melt curves for the three ampoules used for each sample are presented in different colours.
The controls used by participants were as follows: 17 laboratories used in-house materials (previously characterized clinical samples from patients and normal controls), 6 laboratories used DNA samples supplied by the Coriell Institute and 1 lab used NIST PCR product controls.
Tables listing the full genotyping results of the validation study are available as Supplementary Data. Summaries of the clinical interpretations are as follows: samples 1, 11 and 12 (normal female): 19 labs reported ‘normal female’ (or ‘normal genotype’). Exceptions to this interpretation were: lab 11 did not get a PCR product with one out of the three samples, and reported normal female for the other two samples. lab 19 obtained genotypes in all three samples but was unable to complete the investigation and made no interpretation. Samples 2, 7 and 14 (female, pre-mutation): 16 labs reported ‘female, pre-mutation’. Exceptions to this interpretation were: lab 14, which detected the pre-mutation in only one out of three samples and was unable to make an interpretation for two samples; lab 11 would refer all three samples for Southern blotting; labs 15 and 17 reported all three samples as ‘normal female’; lab 19 obtained genotypes in all three samples but was unable to complete the investigation and made no interpretation. Samples 3, 8 and 17 (female, full mutation): 14 labs reported ‘female full mutation’. Exceptions to this interpretation were: lab 16 reported ‘female full mutation’ with only two out of three samples and would refer the third sample for Southern blotting; labs 11, 14 and 15 would refer all samples for Southern blotting; lab 17 reported a different result for all three samples (normal male, male or Turner's syndrome, female full mutation); lab 19 only tested two samples and obtained genotypes but was unable to complete the investigation and made no interpretation; Lab 21 reported only ‘one allele detected by PCR’ and made no clinical interpretation. Samples 4, 10 and 18 (male full mutation): 14 labs reported ‘male full mutation’. Exceptions to this interpretation were: labs 11, 15 and 16 suspected a full mutation and would refer all samples for Southern blotting; lab 8 reported ‘normal male’ with all three samples; lab 17 reported ‘male pre-mutation’ with two out of three samples and ‘male full mutation’ with the third sample; lab 19 was unable to complete the investigation and made no interpretation. Lab 21 did not amplify a PCR product and made no interpretation. Samples 5, 15 and 16 (female, full mutation): 14 labs reported ‘female full mutation’. Exceptions to this interpretation were: lab 8 suspected a full mutation but was unable to obtain Southern blot data; labs 11, 14 and 15 would refer all samples for Southern blotting; lab 17 reported ‘normal female’ with two out of three samples and ‘female full mutation for the third sample; lab 19 was unable to complete the investigation and made no interpretation; lab 21 reported only ‘one allele detected by PCR’. Samples 6, 9 and 13 (male, pre-mutation): 17 labs reported ‘male pre-mutation’. Exceptions to this interpretation were: labs 11 and 15 suspected a pre-mutation and would refer all samples for Southern blotting; lab 17 reported ‘normal male’ for two samples and ‘male full mutation’ for the third sample; lab 19 was unable to complete the investigation and made no interpretation.
A total of 18 non-consensus results were reported, giving an overall rate of non-concordance of 4.9% (21 laboratories × 18 samples – 7 samples not tested), although these were clustered in three laboratories. There was no correlation between the non-concordant results and any particular sample or a specific method. One laboratory reported 12 of the 18 non-concordant results. This laboratory was contacted, and their testing protocol was changed.
Genotyping results are presented in Figure 4. The sample testing mean allele sizes and ranges obtained are summarized in Table 3. Three outliers identified by Grubbs' Test were omitted from the data analysis.19 These were: female full mutation 1 – allele 2: 575 repeats; female full mutation 2 – allele 1: 26 repeats; male full mutation – 7 repeats.

Distribution of allele repeat numbers reported by study participants. The graphs show the number of laboratories (y-axis) detecting each allele length (x-axis). The number of repeats was calculated for each sample in all laboratories, where possible, by taking a mean of all three replicates of the same sample tested in one or more techniques. Where a range of repeats was given the mean value was used. Values reported with ‘greater than’ or ‘less than’ symbols (>, <) were omitted. Where an approximate size was given, this was taken as definitive. All values have been rounded up to whole numbers for clarity. Sample outliers as identified by Grubbs' Test have been omitted.
The detection of a very small 7 repeat allele in the male full mutation samples by two laboratories was unexpected. Such minimal repeats are occasionally detected in full mutation clinical samples and it is unclear if the small repeat is an assay artifact or a true mosaic allele. In this study, one laboratory (lab 8) found the seven repeats by PCR and interpreted the result as a normal male without carrying out Southern blotting, whereas the other laboratory (lab 18) found a similar peak by PCR but noted it as a likely artifact and continued to identify a full expansion by Southern blotting.
Discussion
In the absence of RMs, genetic tests are being performed without adequate controls and new assays are difficult to validate. The need for RMs in Fragile X syndrome testing is particularly pressing, as it is a technically challenging test. For use in in vitro diagnostic assays, RMs in the EU must be CE-marked or in the United States must be FDA-approved. Alternatively, according to the EU IVD directive (Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices), RMs of ‘higher order’ are exempted from CE marking. WHO international standards are accepted as internationally recognized ‘highest order’ measurement standards.
Although aimed to promote the wider availability and safety of commercial products for in vitro diagnostics, the European IVD directive may have hampered RM development and use by preventing the distribution of ‘in-house’ quality control samples between laboratories. This also hampers harmonization of quality between laboratories. Despite the existence of a PCR-product-based certified RM for Fragile X testing (NIST, USA), there was a need for a more commutable, that is, cellular or genomic, certified RM. As EBV-positive cell lines are classified as requiring handling in Category 2 facilities and their shipment must be handled as ‘infectious’, the use of EBV-established cell lines as RMs is not practical. We therefore chose to produce a panel of genomic DNAs derived from cell lines, which could be certified as a ‘higher order’ RM panel. Purified genomic DNA was freeze-dried in glass ampoules. Twenty-one laboratories from seventeen countries collaborated in a validation study of the panel of proposed RMs.
Analysis of material homogeneity showed a significant level of variability between fills and between ampoules. Reasons for this are not clear. That the source of the variation is not due to the spectrophotometer is demonstrated by the low level of variability within ampoules and within the control sample reads (n=10; %CV=1.84). Variation in DNA concentration between fills may be due to inaccurate quantitation before ampoule filling, or due to poor homogeneity of the bulk DNA before filling. Variation between ampoules of the same fill may also be due to poor homogeneity of the bulk DNA. It is difficult to achieve complete homogeneity in solution while retaining the high-molecular-weight DNA required for this application. Because of this variability, users are requested to determine the DNA concentration of each ampoule themselves. The high level of homogeneity within the reconstituted ampoules, however, means that the user can be confident that the sample concentrations will not significantly vary between aliquots from a single ampoule.
Analysis of material stability by Q-PCR and gel electrophoresis of accelerated degradation samples shows some degradation at +56 °C after 2 years. However, the limited extent of degradation at elevated temperature and the absence of degradation at the storage temperature of –20 °C after 2 years indicates that the DNA should be stable for many years. Further samples stored at these temperatures as well as others stored at intermediate temperatures will be monitored at regular intervals in the future as part of planned quality control testing.
DNA profiling of the materials showed very small additional peaks at two loci in one sample (07/174). These peaks were not detected in the original blood samples. That the participants of the collaborative study did not obtain any anomalous results specific to this material and the lack of additional peaks at other loci suggest that these very minor peaks are caused by microsatellite mosaicism rather than contamination.
The wide spread of allele sizes obtained in the collaborative validation study is typical of that obtained in Fragile X testing external quality assessment schemes. A total of 18 incorrect results was reported, giving an overall error rate of 4.9%. This error rate is similar to the 5% level found in an Italian 5-year quality assessment,7 but higher than that observed in our previous studies with different panels (Factor V Leiden: 0.7%; Prothrombin mutation G20210A: 0.7%; Haemophilia A Intron 22 Inversion: 1.8%)20 (also, EG, unpublished data). The high variability of the results, both in reported repeat numbers and the interpretation of results, emphasizes the difficulty of FMR1 repeat sizing and the need for Fragile X syndrome RMs. The finding by two laboratories of an unexpected 7-repeat PCR product is a concern. It is unclear if this small additional putative allele is representative of the clinical sample from which it was derived or is a cell culture or typing artifact. To avoid confusion in the use of this sample as an RM, a caution on the possibility of detecting this putative allele is included in the ‘Instructions for Use’ for the panel. Although the presence of this allele makes the sample unsuitable for use in external quality assessment, it does not detract from the sample's utility as an RM, once the user is aware of it.
By producing the reference panel from a single large batch of pooled DNA derived from a single bulk cell culture batch, the need to repeatedly re-culture the cells for the production of multiple small batches is avoided. This is important as new size alleles can be generated over time in cell culture, particularly when allelic methylation is only partial. It is therefore possible that if this RM batch is ever exhausted, the genotype of the replacement batch may not be identical. However, at that stage a repeat collaborative validation study would be performed to characterize the materials fully.
Two female full mutation samples were included in the study as neither had been previously genotyped and demonstrated to be suitable for inclusion in a reference panel. Both samples performed well. To maintain the widest variety of allele sizes, sample 1 (07/168) was included in the reference panel of five samples (normal female, female pre-mutation, male pre-mutation, female full mutation, male full mutation).
All laboratories that participated in the collaborative study were sent a study report and their approval was sought. All laboratories agreed that the candidate material is suitable for use as a genetic reference panel for Fragile X syndrome. The most frequent comment from the participants (n=6) was that the DNA was of good quality. Three laboratories reported that the DNA concentration varied more than expected and two laboratories did not like the glass ampoules. The report was then endorsed by the board of the European Society for Human Genetics and was finally submitted to the Expert Committee on Biological Standardization at the World Health Organization (WHO). In November 2008, the panel was established as a WHO International Standard and made available through the NIBSC product catalogue (http://www.nibsc.ac.uk/products/catalogue.html) - code: 08/158.
The panel is the only certified set of genomic DNA RMs for Fragile X testing and is available to all diagnostic laboratories worldwide to aid accuracy in Fragile X syndrome testing. The panel will be of particular importance for the validation of commercial diagnostic kits for Fragile X syndrome that are beginning to enter the market, for laboratories setting up new in-house methods such as methylation analysis and for validating existing techniques after a change of reagents, operator or equipment.
References
Sherman S, Pletcher BA, Driscoll DA : Fragile X syndrome: diagnostic and carrier testing. Genet Med 2005; 7: 584–587.
Fu YH, Kuhl DP, Pizzuti A et al: Variation of the CGG repeat at the Fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Kunst CB, Warren ST : Cryptic and polar variation of the Fragile X repeat could result in predisposing normal alleles. Cell 1994; 77: 853–861.
Macpherson JN, Bullman H, Youings SA, Jacobs PA : Insert size and flanking haplotype in Fragile X and normal populations: possible multiple origins for the Fragile X mutation. Hum Mol Genet 1994; 3: 399–405.
Macpherson J, Sawyer H : Practice Guidelines for Molecular Diagnosis of Fragile X Syndrome. Clinical Molecular Genetics Society, 2005, http://cmgsweb.shared.hosting.zen.co.uk/BPGs/pdfs%20current%20bpgs/Fragile%20X.pdf.
Khaniani MS, Kalitsis P, Burgess T, Slater HR : An improved diagnostic PCR assay for identification of cryptic heterozygosity for CGG triplet repeat alleles in the Fragile X Gene (FMR1). Mol Cytogenet 2008; 1: 5.
Falbo V, Floridia G, Tosto F et al: The Italian External Quality Assessment scheme for Fragile X syndrome: the results of a 5-year survey. Genet Test 2008; 12: 279–288.
Ramsden SC, Deans Z, Robinson DO et al: Monitoring standards for molecular genetic testing in the United Kingdom, the Netherlands, and Ireland. Genet Test 2006; 10: 147–156.
Dombrowski C, Lévesque S, Morel ML, Rouillard P, Morgan K, Rousseau F : Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of Fragile X syndrome alleles. Hum Mol Genet 2002; 11: 371–378.
Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F : Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn 2009; 11: 324–329.
Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K : Prevalence of carriers of premutation-size alleles of the FMRI gene—and implications for the population genetics of the Fragile X syndrome. Am J Hum Genet 1995; 57: 1006–1018.
Toledano-Alhadef H, Basel-Vanagaite L, Magal N et al: Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet 2001; 69: 351–360.
Cronister A, Teicher J, Rohlfs EM, Donnenfeld A, Hallam S : Prevalence and instability of Fragile X alleles: implications for offering Fragile X prenatal diagnosis. Obstet Gynecol 2008; 111: 596–601.
Hagerman RJ, Leehey M, Heinrichs W et al: Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of Fragile X. Neurology 2001; 57: 127–130.
Cronister A, Schreiner R, Wittenberger M, Amiri K, Harris K, Hagerman RJ : Heterozygous Fragile X female: historical, physical, cognitive, and cytogenetic features. Am J Med Genet 1991; 38: 269–274.
Amos Wilson J, Pratt VM, Phansalkar A et al: Consensus characterization of 16 FMR1 reference materials: a consortium study. J Mol Diagn 2008; 10: 2–12.
Bonilla V, Sobrino F, Lucas M, Pintado E : Epstein-Barr virus transformation of human lymphoblastoid cells from patients with Fragile X syndrome induces variable changes on CGG repeats size and promoter methylation. Mol Diagn 2003; 7: 163–167.
World Health Organization: Recommendations for the Preparation, Characterization and Establishment of International and Other Biological Reference Standards. World Health Organization Technical Report Series, No. 932. WHO Press: Geneva, 2005.
Grubbs F : Procedures for detecting outlying observations in samples. Technometrics 1969; 11: 1–21.
Gray E, Hawkins JR, Morrison M et al: Establishment of the 1st International Genetic Reference Panel for Factor V Leiden, human gDNA. Thromb Haemost 2006; 96: 215–219.
Acknowledgements
We thank Emma Gillaspy and Jacqueline Rice (Manchester) and James Macpherson (Salisbury) for preliminary testing and field trials. We gratefully acknowledge the following individuals who participated in the collaborative study: Prof C Beldjord, Paris; Dr B Bennetts, Sydney; Dr J Macpherson, Salisbury; Dr M Mila, Barcelona; Dr Z Musova, Prague, Dr E Raukas, Turku; Dr Reena Ray, Dr E Spriggs, Winnipeg; Dr K Richie, Gaithersburg; Dr F Rousseau, Dr J-S Côté, Quebec City; Dr M R Dos Santos, Porto; Dr R Saxena, Dr I C Verma, Dr S Kohli, New Delhi, Prof M Schwartz, Copenhagen; Dr H Slater, Melbourne, Dr I Vlasak, Salzburg; Dr J Weiss, Dr M Losekoot, Leiden; E Doherty, R Williams, A Zhang, C Yang, Auckland; Dr T Lenk, Mr K Young, Celera, USA; Dr M Zannusi, Milan. This work was partly funded by the UK Department of Health through the National Genetics Reference Laboratory (Manchester), an EU FP5 grant ‘CRMGEN’ Contract No. G6RD-2001-00581 and the EU FP6 Network of Excellence EuroGentest’ Contract 512148.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A fee is charged for the panel by NIBSC to cover handling and shipping.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Hawkins, M., Boyle, J., Wright, K. et al. Preparation and validation of the first WHO international genetic reference panel for Fragile X syndrome. Eur J Hum Genet 19, 10–17 (2011). https://doi.org/10.1038/ejhg.2010.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.135
Keywords
This article is cited by
-
Assessment of FMR1 triplet repeats in patients affected with mental retardation, fragile X syndrome and primary ovarian insufficiency
Journal of Genetics (2020)
-
EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders
European Journal of Human Genetics (2015)
-
Establishment of the first WHO international genetic reference panel for Prader Willi and Angelman syndromes
European Journal of Human Genetics (2011)
