
Abstract
17β-Hydroxysteroid dehydrogenase 10 (HSD10) is a mitochondrial enzyme involved in the degradation pathway of isoleucine and branched-chain fatty acids. The gene encoding HSD10, HSD17B10, has been reported as one of the few genes that escapes X-inactivation. We previously studied two female patients with HSD10 deficiency, one of them was severely affected and the other presented a mild phenotype. To elucidate as to why these two carriers were so differently affected, cDNA analyses were performed. The HSD17B10 cDNA of eight control cell lines, two hemizygous patients and two carriers was obtained from cultured fibroblasts, amplified by PCR and sequenced by standard methods. All HSD17B10 cDNAs were quantified by real-time PCR. In the fibroblasts of the female patient who presented with the severe phenotype, only the mutant allele was identified in the cDNA sequence, which was further confirmed by relative quantification (RQ) of HSD17B10 cDNA. This is in agreement with an unfavourable X-inactivation. The other female patient, with slight clinical affectation, showed the presence of both mutant and wild-type alleles in the cDNA sequence, which was confirmed by RQ of HSD17B10 cDNA in fibroblasts. This is in line with normal X-inactivation and the expression of both alleles in different cells (functional mosaicism). RQ results of HSD17B10 cDNA did not differ significantly between male and female controls, which indicate that the genetic doses of mRNA of HSD17B10 was the same in both sexes. In conclusion, these results suggest that the HSD17B10 gene does not escape X-inactivation as has been reported previously.
Similar content being viewed by others
INTRODUCTION
17β-Hydroxysteroid dehydrogenase 10 (HSD10) is a mitochondrial enzyme involved in the degradation pathway of isoleucine and branched-chain fatty acids.1 This enzyme has also been found to be involved in the metabolism of sex steroid hormones, neuroactive steroids and in the detoxification of cytotoxic aldehydes.2, 3 HSD10 deficiency (OMIM 300256) is an X-linked defect caused by mutations in the HSD17B10 gene. Clinically, the great majority of male patients show normal early development followed by progressive loss of mental and motor skills.1, 4, 5, 6, 7, 8, 9, 10, 11 However, three patients were identified who presented symptoms in the first days of life.1, 11 It has recently been shown that symptoms of these patients are unrelated to accumulation of metabolites in the isoleucine pathway and that the neurological handicap can be associated with an imbalance in neurosteroid metabolism12 or to defects in general mitochondrial function.13 In addition, the splice variant c.574C>A of HSD17B10 gene has been associated with a new syndromic form of X-linked mental retardation, choreoathetosis and abnormal behaviour.14
The HSD17B10 gene has been mapped to chromosome Xp11.215 and has been reported as one of the few genes that escapes X-inactivation.16 To date, 10 female patients with HSD10 deficiency have been described presenting a variety of symptoms, from borderline learning difficulties to psychomotor and speech delay.5, 9, 11 We previously studied two of these female patients. One of them was heterozygous for the p.N247S mutation and was severely affected, whereas the other was heterozygous for the p.P210S mutation and presented a slight clinical affectation.11 To elucidate as to why these two female patients were so differently affected, we performed HSD17B10 cDNA quantitative analysis in both female patients and in control fibroblasts, the results of which are reported here.
MATERIALS AND METHODS
Material
Skin biopsies from patients of two unrelated Spanish families with HSD10 deficiency were obtained: family 1 consisting of a male patient (1IIM) and his carrier sister (1IIF), both with a severe phenotype (Figure 1a); family 2 consisting of a male patient (2IIM), with a severe phenotype, and his heterozygous mother (2IF), with a slight clinical affectation (Figure 1a). Both families have been described previously.11 Eight cell lines (four males and four females) from our cell bank were used as controls.

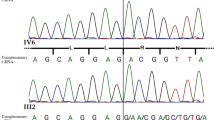
Pedigree (a) and cDNA sequence (b) of families 1 and 2.
All the samples were obtained according to the Declaration of Helsinki and informed consent was signed by all the patients or their parents.
Molecular studies
cDNAs were obtained from cultured fibroblasts, were amplified by PCR and sequenced using standard protocols and oligonucleotides designed in-house (sequences available upon request). All HSD17B10 cDNAs were quantified by the StepOnePlus real-time PCR System using the Comparative Ct (ΔΔCt) method from StepOne software v2.0 (Applied Biosystems, Foster City, CA, USA). The Primer Express 3.0 software (Applied Biosystems) was used to design two sets of primers and probes to differentiate wild-type (Wt) and mutant (Mut) alleles corresponding to mutations p.N247S (c.740A>G) and p.P210S (c.628C>T) of the HSD17B10 gene. We used two different endogenous controls: glyceraldehyde 3-phosphate dehydrogenase (PN4310884E) and cyclophilin A (PPIA, PN4310883E) (Applied Biosystems). As additional control, a mixed pool of four healthy male cDNAs was used in each analysis.
Gene nucleotide numbering was carried out according to sequence RefSeq NM_004493 with +1 as A of the ATG start codon. The ATG codon represents +1 for the amino-acid numbering according to the HSD10 protein sequence NP_004484.
X-inactivation studies
The androgen-receptor locus (AR) methylation assay was performed in genomic DNA of female carriers as described previously.17 If the AR locus was uninformative, skewing was assessed at FMR1 locus.18 Briefly, genomic DNA (300 ng) was digested with 5 U of HpaII (both for the AR and FMR1 assays) in a total volume of 20 μl. For each sample, an undigested control was prepared. We define the pattern of X-chromosome inactivation as skewed when the inactivation percentage was over 80%.
Statistical methods
Statistical studies for the analyses of relative quantification (RQ) in male and female controls were performed using the non-parametric two-related sample Wilcoxon's test, with the SPSS software (version 14.0 for Windows).
RESULTS AND DISCUSSION
HSD17B10 has been reported as one of the few genes that escapes X-inactivation,16 which predicts that female carriers would not be affected. However, 10 female carriers with HSD10 deficiency have been described so far, presenting different degrees of clinical affectation, which is in agreement with an X-linked inheritance with different degrees of X-inactivation.11
To elucidate whether HSD17B10 cDNA doses differed between both sexes, we performed RQ of Wt HSD17B10 cDNA alleles in four female and four male controls (Figure 2). Results of the Wilcoxon statistical test did not show any significant difference between the doses in both sexes, considering the two endogenous controls (P-value=0.07). Therefore, these results are in favour of an X-linked disease that does not escape X-inactivation.

RQ of HSD17B10 Wt probes named as p.N247N and p.P210P, with two distinct endogenous controls (glyceraldehyde 3-phosphate dehydrogenase and cyclophilin A) in four male and four female controls. The bars represent the mean of four controls and the error bars represent the mean±SD.
We previously studied two unrelated female patients with different degrees of clinical affectation.11 The female of family 1 (1IIF), like her brother (1IIM), presented a severe phenotype with psychomotor and speech delay, and a clear deficiency of HSD10 activity in fibroblasts.11 When we sequenced her HSD17B10 cDNA, it seemed that only the Mut allele was identified (Figure 1a). Results for the RQ of her HSD17B10 cDNA (Figure 3a) showed that amplification levels of the Mut probe were much higher than those of the Wt probe and very similar to those of her brother (1IIM), independently of the endogenous control used (Figure 3a). To rule out a Turner syndrome, chromosome analysis was performed, which resulted in a normal karyotype (46,XX). Skewed X-inactivation was confirmed by methylation studies. Patient 1IIF was homozygous for AR locus, consequently this study was uninformative, but FMR1 locus showed a skewed X-inactivation pattern (80/20). These results are in agreement with an unfavourable X-inactivation effect of HSD17B10 gene in the analysed tissue. In addition, as the girl was severely affected, a similar unfavourable X-inactivation in other tissues could be expected.

RQ of HSD17B10 Wt and Mut probes with two distinct endogenous controls (glyceraldehyde 3-phosphate dehydrogenase and cyclophilin A) in patient and control cDNAs (a) family 1, (b) familiy 2. The bars of controls represent the mean of eight controls (four males+four females), performed in triplicate. The bars of patients represent the mean of triplicate measurements. The error bars represent the mean±SD.
The other female patient (2IF) showed a mild clinical affectation, with learning disabilities and HSD10 activity in fibroblasts within the control range.11 HSD17B10 cDNA sequencing showed the presence of both Mut and Wt alleles (Figure 1b). This observation was in agreement with the results of the RQ studies showing similar HSD17B10 cDNA levels of both Wt and Mut probes, while we were only able to amplify the Mut probe in her severely affected son (2IIM) (Figure 3b). X-inactivation analysis showed a random X-inactivation pattern for AR locus in patient 2IF. These results suggest the presence of both HSD17B10 alleles in this female patient, which is in line with normal X-inactivation and the expression of both alleles in different cells (functional mosaicism). In addition, the normal enzymatic activity found in this female patient11 might be due to lack of sensitivity of the enzymatic technique, or maybe there was enough dose of Wt HSD17B10 mRNA to produce enough HSD10 protein to obtain normal activity.
However, we did observe that the amplification responses were different for each probe when they were corrected by the two different endogenous controls (Figure 3). This could be explained by the low specificity of the probes and by the variability of the endogenous controls. However, in spite of it, the interpretation of the results did not change.
To summarise, here we present the results of HSD17B10 cDNA analysis in two female carriers compared with two male patients and controls. The hypothesis that HSD17B10 is inactivated in one of the X-chromosome is supported by the results in controls, which showed that doses of HSD17B10 cDNA were the same in both sexes (Figure 2). RQ cDNA results for one of the female patient (1IIF), together with the enzymatic studies and the severe clinical presentation, were in agreement with an unfavourable X-inactivation effect. In addition, RQ cDNA results for the other female patient (2IF) seem to reflect the presence of a mosaicism in the studied tissue, which could explain the normal enzymatic activity and her mild phenotype. However, we cannot exclude that differences in disease severity between both female carriers is at least partly due to differences in the effect of the mutations, as the male patient with the p.N247S mutation died at age 2 months, while the patient with p.P210S mutation is alive at 4 years of age.
In conclusion, our results suggest that HSD17B10 gene does not escape X-inactivation as reported previously.16 Heterozygous female patients showed the classical biochemical and clinical variability of X-linked diseases due to random X-chromosome inactivation and the severity of the phenotype will depend on the total dose of Mut mRNA in different tissues as well as on the severity of the mutation.
References
Zschocke J, Ruiter JPN, Brand J et al: Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr Res 2000; 48: 852–855.
Yang SY, He XY, Schulz H : Multiple functions of type 10 17β-hydroxysteroid dehydrogenase. Trends Endocrinol Metab 2005; 16: 167–175.
Murakami Y, Ohsawa I, Kasahara T, Ohta S : Cytoprotective role of mitochondrial amyloid β peptide-binding alcohol dehydrogenase against a cytotoxic aldehyde. Neurobiol Aging 2009; 30: 325–329.
Poll-The BT, Wanders RJA, Ruiter JPN et al: Spastic diplegia and periventricular white matter abnormalities in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency, a defect of isoleucine metabolism: differential diagnosis with hypoxic–ischemic brain diseases. Mol Genet Metab 2004; 81: 295–299.
Ensenauer R, Niederhoff H, Ruiter JPN et al: Clinical variability in 3-hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. Ann Neurol 2002; 51: 656–659.
Olpin SE, Pollitt RJ, McMenamin J et al: 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency in a 23 year old man. J Inherit Metab Dis 2002; 25: 477–482.
Sass JO, Forstner R, Sperl W : 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: impaired catabolism of isoleucine presenting as neurodegenerative disease. Brain Dev 2004; 26: 12–14.
Sutton VR, O'Brien WE, Clark GD, Kim J, Wanders RJA : 3-Hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2003; 26: 69–71.
Perez-Cerdá C, García-Villoria J, Ofman R et al: 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: an X-linked inborn error of isoleucine metabolism that may mimic a mitochondrial disease. Pediatr Res 2005; 58: 488–491.
Cazorla MR, Verdú A, Pérez-Cerdá C, Ribes A : Neuroimage findings in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency. Pediatr Neurol 2007; 36: 264–267.
García-Villoria J, Navarro-Sastre A, Fons C et al: Study of patients and carriers with 2-methyl-3-hydroxybutyryl-CoA deshydrogenase (MHBD) deficiency: difficulties in the diagnosis. Clin Biochem 2009; 42: 27–33.
Yang S-Y, He X-Y, Olpin SE et al: Mental retardation linked to mutations in the HSD17B10 gene interfering with neurosteroid and isoleucine metabolism. Proc Natl Acad Sci USA 2009; 106: 14820–14824.
Rauschenberger K, Schöler K, Sass JO, Sauer S, Djuric Z : A non-enzymatic function of 17β-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol Med 2010; 2: 1–12.
Lenski C, Kooy RF, Reyniers E et al: The reduced expression of the HADH2 protein causes X-linked mental retardation, choreoathetosis, and abnormal behaviour. Am J Hum Genet 2007; 80: 372–377.
He X-Y, Schulz H, Yang S-Y : A human brain L-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid beta-peptide-binding protein involved in Alzheimer's disease. J Biol Chem 1998; 273: 10741–10746.
Miller AP, Willard HF : Chromosomal basis of X chromosome inactivation: identification of a multigene domain in Xp11.21–p11.22 that escapes X inactivation. Genetics 1998; 95: 8709–8714.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW : Methylation of HpaII and HhaI sites near polymorphic CAG repeat in the human androgen-receptor gene correlates with X-chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Müller A et al: Expand long PCR for fragile X mutation detection. Clin Genet 1997; 52: 147–154.
Acknowledgements
We thank Carlota Ogg and Patricia Alcalá for their excellent technical assistance. This group is funded by the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII and CENIT 06/Nanopharm. Cristina Fernández is a recipient of a grant (FIS, CA 06-0128). This work has been performed in the context of the PhD programme of the Department of Biochemistry and Molecular Biology of the University of Barcelona.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
García-Villoria, J., Gort, L., Madrigal, I. et al. X-inactivation of HSD17B10 revealed by cDNA analysis in two female patients with 17β-hydroxysteroid dehydrogenase 10 deficiency. Eur J Hum Genet 18, 1353–1355 (2010). https://doi.org/10.1038/ejhg.2010.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.118
Keywords
This article is cited by
-
The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation
Journal of Human Genetics (2014)
-
Transcription start sites and epigenetic analysis of the HSD17B10 proximal promoter
BMC Biochemistry (2013)
-
HSD10 disease: clinical consequences of mutations in the HSD17B10 gene
Journal of Inherited Metabolic Disease (2012)
