
Abstract
Maroteaux–Lamy syndrome is an autosomal-recessive disorder due to the deficit of the lysosomal enzyme, arylsulfatase B (ARSB). Among the numerous genomic lesions reported till now, the sequence variant, c.1151G>A (p.S384N), has been associated with a severe phenotype in more than 10% of the patients. We now report the first in vivo demonstration of the polymorphic nature of p.S384N, revealed during the segregation analysis in a family at risk for Maroteaux–Lamy syndrome. The proband, compound heterozygous for c.[944G>A]+[245T>G] (p.[R315Q]+[L82R]), did not carry the p.S384N change, which was instead present in two healthy members of the family, in trans with the causative mutations, p.R315Q and p.L82R, respectively. The hypothesis that p.S384N was a polymorphism was further addressed by reverse dot-blot analysis of 400 control alleles, estimating an allele frequency of 4.5%. To predict the consequences of p.R315Q, p.L82R and p.S384N, we also modeled and compared the three amino-acid changes in the three-dimensional ARSB structure. The in silico analysis predicted a local protein misfolding in the presence of p.R315Q and p.L82R. On the contrary, no evident problem was predicted in the case of p.S384N, occurring on the protein surface, far from the active site. Overall, these findings strongly support the hypothesis that the non-synonymous change p.S384N is a polymorphism. Moreover, our results emphasize the need for caution in drawing conclusions from a novel variant allele before screening at least 50 healthy control subjects.
Similar content being viewed by others

Introduction
Maroteaux–Lamy, or mucopolysaccharidosis Type VI (MPSVI, MIM #253200), is an autosomal-recessive disorder due to the lysosomal defect of arysulfatase B (ARSB, EC 3.1.6.1). Its deficiency causes a storage of dermatan sulfate resulting in a wide variety of clinical phenotypes, including mainly hepatosplenomegaly, corneal clouding, growth retardation and skeletal deformities.1 The incidence of the disorder is quite low and varies in different areas of the world, the highest being 1:248 000 live births in Australia.2 The ARSB gene, located in 5q11-13, spans a region of about 206 kb and contains eight exons. More than 100 different alterations have been described, mainly represented by point mutations.3, 4 In addition, few polymorphisms have been identified in the gene sequence. The substitution c.1151G>A (p.S384N) has been controversially reported as polymorphism5, 6 or, more frequently, as common pathogenic mutation.3, 7
In particular, Karageorgos et al7 reported p.S384N as deleterious mutation in 12.4% of the patients. Hence, the presence of the p.S384N in trans with the known disease-causing mutations, c.944G>A (p.R315Q)8 or c.245T>G (p.L82R),5 in two healthy relatives of an affected patient, prompted us to further investigate whether the serine replacement to asparagine in position 384 was indeed a pathological lesion. The methodological approach was based on reverse dot-blot (RDB) analysis of 400 alleles in the normal Italian population followed by direct sequencing of the positive samples. In addition, a three-dimensional ARSB structure model was used to predict the molecular consequences of the amino-acid changes.
Materials and methods
Subjects analyzed
The patient, first child of healthy non-consanguineous parents, showed at 4 months of age typical symptoms and signs of MPS (kyphosis, coarse facies, hepatosplenomegaly and mitral valve regurgitation). At 10 months, the diagnosis of MPSVI was completed through the demonstration of increased urinary glycosaminoglycans and reduced ARSB activity in white blood cells (5.2 nmol/mg/h; normal value 129.3±33 nmol/mg/h; assay according to Baum et al9). She then developed short stature, other skeletal deformities, joint contractures, hearing impairment and corneal clouding. She started enzyme replacement therapy at 4.3 years of age.
Blood samples were obtained from the MPSVI patient and her relatives upon informed consent and from the 200 volunteer control subjects anonymously.
ARSB mutation analysis
Genomic DNA was extracted from peripheral blood leukocytes using the commercial QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). ARSB exons and their flanking regions were PCR-amplified using appropriate intronic primers designed by Primer 3 software (http://fokker.wi.mit.edu/primer3/input.htm) (sequences and PCR conditions available upon request). Duplicate PCR products were sequenced with the ABI PRISM BigDye Terminator Cycle Sequencing Kit (Applied Biosystems).
RDB analysis
PCR amplification of exon 6 for RDB analysis was carried out in a 20 μl volume containing 5 μ M biotin-16-dUTP (Roche, Mannheim, Germany). A specific set of primers was designed for amplification of exon 6 (forward: 5′-AACTGGCAGGTTTGTTATTTCC-3′, reverse: 5′-GCAACTCAGGAAAAAAGAGAAAGC-3′) and supplied by Sigma Genosys.
Wild-type and mutant allele-specific oligonucleotide probes were designed for the variant p.S384N and for the known polymorphism c.1191G>A (p.P397P) (Table 1).
Probes were covalently bound to the membrane: each membrane strip was dotted with the normal (N) and the mutant (M) probes for the p.S384N and p.P397P sequence variants. Probes binding, PCR products hybridization and detection were performed as described earlier.10
The confirmation of the variants detected by RDB was obtained by sequencing the purified exon 6 PCR product, as described above.
Mutation nomenclature
Mutations are described according to the nomenclature, numbering nucleotide +1 the A of the first ATG translation initiation codon (http://www.hgvs.org/mutnomen).11, 12 Nucleotide numbers are derived from cDNA sequence (EMBL/GenBank/DDBJ; accession no. M32373.1) and from genomic sequence (NC_000005.8).
Model building
The three-dimensional structure of human lysosomal ARSB determined at 2.1 Å resolution by crystallographic studies (PDB code 1FSU) has been used as a template for the mutational studies. Site-specific residue substitutions have been carried out using the program O, and the obtained models for mutated proteins were energy-minimized using the program Discover 3 from the Insight II suite (Accelrys–MSI, San Diego, CA, USA). Figure 1 was drawn using the program CHIMERA.13, 14, 15

Modeling the three amino-acid changes into the three-dimensional structure of the full-length arylsulfatase B. (a) Hydrophobic cluster (white residues) in proximity of L82 (yellow). The long-branched side chain of the replacing arginine R82 (green) may cause clashes between atoms and hydrophobic cluster disruption. (b) Hydrogen bond network built by residue R315 (yellow), four water (WAT) molecules, D466 and the OH group of E483 (white residues). Residues W322 and W450 encompass the side chain of the wild-type R315 by forming a sort of stacking interaction. (c) Residue S384 (drawn in yellow as space-filling model) is fully exposed to the solvent on the protein surface, far from the active site area.
Results and discussion
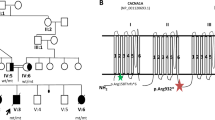
We here report the first in vivo demonstration of the polymorphic nature of the variant p. S384N that was revealed during the segregation analysis in a family at risk for Maroteaux–Lamy syndrome. Results are reported in Figure 2.

Pedigree of the informative MPSVI family. Circle, females; square, males; black symbol, affected proband; open symbols with solid symbol inside, carrier.
The proband, compound heterozygous for the causative mutations c.944G>A (p.R315Q)8 and c.245T>G (p.L82R),5 also carried the known allelic synonymous change p.P397P in homozygosis. Segregation analysis showed that the p.R315Q mutation was of paternal origin, and confirmed the mother as carrier of the p.L82R mutation. As expected, both parents also carried the polymorphism p.P397P. Sequence analysis revealed that the father, in addition to the mutation p.R315Q and the polymorphism p.P397P, also carried the p.S384N variant. Although in all the earlier reports p.R315Q and p.S384N were always found in cis on the same allele,7, 16 the evidence that the amino-acid change (p.S384N) was not detected in the patient led us to consider the hypothesis that p.R315Q and p.S384N were in trans in the apparently healthy father. This was confirmed by the extension of the segregation analysis to a healthy sister of the patient, who resulted to be the carrier of the maternal c.245T>G (p.L82R) allele, in association with the paternal c.1151 G>A (p.S384N) allele.
The presence of the p.S384N variation in trans with respect to both causative mutations gave us, for the first time, the opportunity to perform both clinical and biochemical evaluations on the effect produced by the p.S384N variation.
A careful clinical examination of the father confirmed no MPSVI signs or symptoms. Accordingly, biochemical evaluation of ARSB activity on samples of both the father and the sister detected intermediate values, 54 and 51.5%, respectively, compared with a normal control, as expected for heterozygous subjects. Therefore, our in vivo findings of enzymatic activity, resulting from the contribution of the p.S384N in association with the causative p.L82R or p.R315Q, differ from the in vitro expression results reported by Garrido et al.5 Although absolutely aware of the possible polymorphic nature of p.S384N, the authors could not be conclusive regarding the result of a reduced ARSB activity (57%) detected when p.S384N was analyzed as a single change or of a further ARSB reduction (up to 16%) if p.S384N was analyzed in a double mutant.5 Indeed, the influence of several factors on the expression pattern of pathogenic variants or polymorphisms has to be always taken into account.
Finally, to evaluate the frequency of c.1151G>A (p.S384N) in the Italian population, we analyzed 200 DNA samples (400 alleles) from healthy individuals with RDB technique (Figure 3), including the polymorphism p.P397P as a positive control. We found 18 individuals carrying the p.S384N and 76 subjects having the p.P397P, both variants in the heterozygous state. Four DNA samples showed both the genetic variants. Exon 6 of the p.S384N-positive samples was completely sequenced and the allelic variant confirmed in all 18 cases. Therefore, p.S384N and p.P397P were detected in 9 and 38% of the screened individuals, respectively. If we considered the p.S384N variant as a pathogenetic mutation, its sole allele frequency (estimated to be 4.5% in our population) would be higher than that reported for the entire set of pathogenetic mutations leading to cystic fibrosis (about 4%),17 the most common genetic disorder. To conclude, RDB findings clearly show that the c.1151G>A (p.S384N) is a non-pathogenetic single nucleotide polymorphism.

Reverse dot-blot analysis. Each membrane strip was spotted with the normal (N) and the mutant (M) probe for the p.S384N and the p.P397P gene variants and hybridized to amplified DNA from exon 6 of the gene ARSB. The color was developed with streptavidin-HRP and TMB. The genotype of each individual is shown on the right. A strip with no probe spotted was used as blank. 1=sample; 2=p.S384N; 3=p.P379P; and 4=blank.
Studies on structural models, based on diverse bioinformatics approaches, have been reported earlier.5, 18, 19 In this study, the consequences of p.R315Q, p.L82R and p.S384N were in silico predicted through visual inspection of the three-dimensional structure of the full-length ARSB in terms of both function and spatial orientation of specific residues, as well as disturbance generated by amino-acid substitutions (Figure 1). As shown in Figure 3a, L82 is positioned deep inside the protein core surrounded by the hydrophobic moieties, V80, I296, V332 and I348, and the aromatic rings of Y85 and W353. The substitution of a leucine, with the long-branched side chain, with an arginine may potentially cause destabilizing effects resulting from steric clashes between nearby atoms as well as from the rupture of the hydrophobic cluster. Hence, the mutation L82R is predicted to lead to a local protein misfolding. Moreover, the new 82R residue would lie in close contact with I296, which stands on the same strand and in a short distance from the active site residue D300. This ‘bumping’ could move the strand from the canonic positioning preventing D300 to correctly bind the substrate and/or the Ca2+ ion in the active site. Figure 3b shows the possible consequence of the replacement R315Q. This mutation occurs in close proximity to protein surface but still in a not-fully solvent exposed area. The residue R315 belongs to a hydrogen bond networks binding four water molecules (WAT26, WAT139, WAT185 and WAT209), D466 and the OH group of E483. Moreover, the guanidine group of the arginine side chain is encompassed by the aromatic rings of residues W322 and W450, in a sort of stacking interaction. The side chain of the replacing glutamine (315Q) is shorter and thus loses the binding to D466 and to the water molecules, thereby opening a cavity in proximity of F475 (data not shown). It is likely that this mutation leads to a local misfolding of the protein. Finally, Figure 3c clearly shows as the substitution S384N is not predicted to cause substantial consequence. Both serine (S) and asparagine (N) are polar residues. Despite the resulting protein variant has an amino acid (384N) with a longer side chain, this change, occurring on the protein surface, is fully exposed to the solvent. Hence, there is no evident problem to the protein three-dimensional structure. Our findings support those obtained in an earlier study by Saito et al.19 The authors, by using a different bioinformatics approach, had reported in silico findings not in line with the severe phenotype that, in large cohort studies, had frequently been associated with the S384N substitution. To date, about 10% of the MPSVI cases have been reported as attributable to the p.S384N variant mostly in the heterozygous state, but also in homozygosis.7
Hence, on the basis of our results, the disease-causing mutations in these patients is still unidentified, thus suggesting that a considerable number of mutations could be located out of the coding sequence or could be represented by partial deletions or might be simply missed.
These findings illustrate the importance of a comprehensive evaluation, including segregation analysis and full sequencing, when characterizing a family at risk. The misidentification of a non-pathogenetic polymorphism as mutation has important implications related not only to pre- and postnatal diagnoses (especially if these are based on molecular analysis) but also to genetic counseling in the family at risk, because of carrier misdetection. Finally, our results further emphasize the need for caution in drawing conclusions from a novel variant allele before screening at least 50 healthy control subjects.
Accession codes
References
Neufeld E, Muenzer J : The mucopolysaccharidoses; in Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease. 8th edn. McGraw-Hill Medical Publishing Division: New York, 2001, pp 3421–3452.
Brooks DA, Gibson GJ, Karageorgos L, Hein LK, Robertson EF, Hopwood JJ : An index case for the attenuated end of the mucopolysaccharidosis type VI clinical spectrum. Mol Genet Metab 2005; 85: 236–238.
Lin WD, Lin SP, Wang CH et al: Genetic analysis of mucopolysaccharidosis type VI in Taiwanese patients. Clin Chim Acta 2008; 394: 89–93.
Human Gene Mutation Database. Available at: http://www.hgmd.cf.ac.uk.
Garrido E, Cormand B, Hopwood JJ, Chabás A, Grinberg D, Vilageliu L : Maroteaux–Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol Genet Metab 2008; 94: 305–312.
Thorisson GA, Smith AV, Krishnan L, Stein LD : The International HapMap Project Web site. Genome Res 2005; 15: 1591–1593.
Karageorgos L, Brooks DA, Pollard A et al: Mutational analysis of 105 mucopolysaccahridosi type VI patients. Hum Mutat 2007; 28: 897–903.
Villani GR, Balzano N, Vitale D, Saviano M, Pavone V, Di Natale P : Maroteaux–Lamy syndrome: five novel mutations and their structural localization. Biochim Biophys Acta 1999; 1453: 185–192.
Baum H, Dodgson KS, Spencer B : The assay of arylsulphatases A and B in human urine. Clin Chim Acta 1959; 4: 453–455.
Chehab FF, Wall J : Detection of multiple cystic fibrosis mutations by reverse dot blot hybridization: a technology for carrier screening. Hum Genet 1992; 89: 163–168.
den Dunnen JT, Antonarakis SE : Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15: 7–12.
den Dunnen JT, Paalman MH : Standardizing mutation nomenclature: why bother? Hum Mutat 2003; 22: 181–182.
Bond CS, Clements PR, Ashby SJ et al: Structure of a human lysosomal sulfatase. Structure 1997; 5: 277–289.
Jones TA, Zou JY, Cowan SW, Kjeldgaard M : Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 1991; 47: 110–119.
Pettersen EF, Goddard TD, Huang CC et al: UCSF chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Garrido E, Chabás A, Coll MJ et al: Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux–Lamy syndrome) patients, including 9 novel mutations. Mol Genet Metab 2007; 92: 122–130.
Lakeman P, Gille JJ, Dankert-Roelse JE et al: CFTR mutations in Turkish and North African cystic fibrosis patients in Europe: implications for screening. Genet Test 2008; 12: 25–35.
Litjens T, Hopwood JJ : Mucopolysaccharidosis type VI: structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat 2008; 18: 282–295.
Saito S, Ohno K, Sugawara K, Sakuraba H : Structural and clinical implications of amino acid substitutions in N-acetylgalactosamine-4-sulfatase: insight into mucopolysaccharidosis type VI. Mol Genet Metab 2008; 93: 419–425.
Acknowledgements
This work was partly funded by the Italian MPS Association. Some samples were obtained from the ‘Cell Line and DNA Biobank from Patients Affected by Genetic Diseases’ (G Gaslini Institute) – Telethon Genetic Biobank Network (Project no. GTB07001A). We thank Fondazione Pierfranco and Luisa Mariani, Milano for their generous funding of the clinical activity in Monza.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zanetti, A., Ferraresi, E., Picci, L. et al. Segregation analysis in a family at risk for the Maroteaux–Lamy syndrome conclusively reveals c.1151G>A (p.S384N) as to be a polymorphism. Eur J Hum Genet 17, 1160–1164 (2009). https://doi.org/10.1038/ejhg.2009.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.19
Keywords
This article is cited by
-
Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know
Italian Journal of Pediatrics (2018)
-
Lysosomal storage disorders: Molecular basis and laboratory testing
Human Genomics (2011)
-
Mucopolysaccharidosis VI
Orphanet Journal of Rare Diseases (2010)
-
The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes
Nature Genetics (2010)
