
Abstract
In the Victorian era, Sir Francis Galton showed that ‘when dealing with the transmission of stature from parents to children, the average height of the two parents, … is all we need care to know about them’ (1886). One hundred and twenty-two years after Galton's work was published, 54 loci showing strong statistical evidence for association to human height were described, providing us with potential genomic means of human height prediction. In a population-based study of 5748 people, we find that a 54-loci genomic profile explained 4–6% of the sex- and age-adjusted height variance, and had limited ability to discriminate tall/short people, as characterized by the area under the receiver-operating characteristic curve (AUC). In a family-based study of 550 people, with both parents having height measurements, we find that the Galtonian mid-parental prediction method explained 40% of the sex- and age-adjusted height variance, and showed high discriminative accuracy. We have also explored how much variance a genomic profile should explain to reach certain AUC values. For highly heritable traits such as height, we conclude that in applications in which parental phenotypic information is available (eg, medicine), the Victorian Galton's method will long stay unsurpassed, in terms of both discriminative accuracy and costs. For less heritable traits, and in situations in which parental information is not available (eg, forensics), genomic methods may provide an alternative, given that the variants determining an essential proportion of the trait's variation can be identified.
Similar content being viewed by others

Introduction
Height is a classical example of an inherited human trait. More than a 100 years ago, Francis Galton used height data to study the resemblance between parents and offspring, concluding that ‘when dealing with the transmission of stature from parents to children, the average height of the two parents, … is all we need care to know about them’1 (Figure 1). Later on, height was among the first phenotypes studied using the polygenic model of inheritance,2 which bridged the gap between Galtonian and Mendelian genetics. Numerous studies after the pioneering work of Galton showed that height is one of the most heritable human phenotypes. Typically, the proportion of the sex- and age-adjusted variance of height attributable to familial factors (heritability) is estimated as 80%. Most of this heritability may be owing to genetic factors because, for height, the non-genetic causes of sib resemblance are usually negligibly small.3 Until recently, however, little was known about the genes involved in the normal variation of height in human populations.

Rate of regression in hereditary stature (Plate IX, figure a from Galton1 with superimposed data from the ERF study).
One hundred and twenty-two years after Galton’s paper, and 7 years after the initial sequencing of the human genome,4 three papers described 54 loci showing strong statistical evidence for association with height,5, 6, 7 potentially providing us with genomic means of human height prediction. Here, we investigate the potential of the state-of-the-art genomic approach to predict human height and compare it with the potential of the 122-year-old Victorian method of Galton.
Materials and methods
Study populations
The Rotterdam Study8 is a prospective cohort study that started in 1990 in Ommoord, a suburb of Rotterdam, among 10 994 men and women aged 55 and over. The main objective of the Rotterdam Study is to investigate the prevalence and incidence of and risk factors for cardiovascular, neurological, locomotor and ophthalmological diseases in the elderly. Baseline measurements were obtained between 1990 and 1993. All participants were subsequently examined in follow-up examination rounds every 2–3 years. Heights were measured at baseline. The Rotterdam Study has been approved by the institutional review board (Medical Ethics Committee) of the Erasmus Medical Center and by the review board of the Netherlands Ministry of Health, Welfare and Sports. For this study, we used the data on 5748 participants for whom GWA and height data were available.
The Erasmus Rucphen Family (ERF) study9 is a family-based study of a young genetically isolated population studied within Genetic Research in the Isolated Populations program.10 The ERF study includes over 3000 participants descending from 22 couples living in the Rucphen region in the nineteenth century. All descendants were invited to visit the clinical research center in the region, where they were examined in person, including height measurements. The ERF study has been approved by the Medical Ethics Committee of the Erasmus MC. In this study we included 550 participants together with both parents for whom height measurements were complete.
Genotyping and imputations
In the Rotterdam Study, genome-wide SNP genotyping was performed using Infinium II assay on the HumanHap550 Genotyping BeadChips (Illumina Inc., San Diego, CA, USA).
Approximately 2.5 million SNPs were imputed using the HapMap CEU population (release 22) as reference. The imputations were performed using MACH software11 (http://www.sph.umich.edu/csg/abecasis/MACH/). The quality of imputations was checked by contracting imputed and actual genotypes at 78 844 SNPs not present on Illumina 550K for 437 individuals for whom these SNPs were directly typed using Affymetrix 500K. Using the ‘best guess’ genotype for imputed SNPs, the concordance rate was 99% for SNPs with the R2 (ratio of the variance of imputed genotypes to the binomial variance) quality measure greater than 0.9; concordance was still high (94%) when R2 was between 0.5 and 0.9. Out of the 54 SNPs used in this study, 31 were directly typed and the rest were imputed. The median R2 was 0.999 and only two SNPs had 0.87<R2<0.9 (Supplementary Table 1).
In the ERF study, genome-wide SNP genotyping was performed using Illumina HumanHap300 (1200 individuals), HumanHap370 (100 individuals) and Affymetrix 250K Nsp array (∼200 individuals). The imputations followed the Rotterdam Study protocol closely.
Selection of 54 SNPs used in the study
Of the 54 loci influencing human height shown in Supplementary Table 1, 16 were published by Weedon et al.,5 11 by Lettre et al.6 and 27 by Gudbjartsson et al.7 Of the 59 markers reported to be strongly associated with height in these three studies, five were mapped within the same chromosome region. For these loci, we picked up markers with the lowest P-value.
Testing within- and between-loci additivity
All analyses were performed using R v 2.7.0 (http://www.r-project.org). To test the deviation from the within-locus additive model, we used the linear model height ∼βs sex+βa age+βAB PAB+βBB PBB, where PAB and PBB are the estimated probabilities of the AB and BB genotypes, respectively. This model was contrasted to the model under additive restriction βBB=2 βAB using the likelihood ratio test (LRT; twice the difference between maximum log-likelihood of these models is asymptotically distributed as χ12). Multiple testing was accounted for using Bonferroni correction. Similarly, we tested the deviation from between-loci additivity using the model height ∼βs sex+βa age+βS1 DS1+βS2 DS2+βI DS1 DS2, in which DS1 and DS2 were the estimated allele doses at two loci (D=PAB+2PBB) and βI is the interaction term. This model was contrasted to the no interaction model (height ∼βs sex+βa age+βS1 DS1+βS2 DS2); again, LRT on one degree of freedom was performed for comparison.
Construction of predictive profiles
The non-weighted allelic profile was computed as the sum of the estimated doses of the height-increasing allele in the genotype of a person. The weighted allelic profile was constructed as a weighted allelic sum with weight proportional to the allelic effect estimated using our data in a multivariable model including all 54 SNPs.
To construct the Galtonian mid-parental profile, we first estimated height residuals from the model height ∼sex+age. For every person for whom both paternal and maternal heights were available, we constructed the ‘predictive profile’, which was defined as the average of the parental height residuals. This method resembles the method of Galton, very closely1 with the exception that he did not adjust for age.
The hypothetical predictor explaining a certain proportion (Ve) of sex- and age-adjusted height variance was constructed as a sum of the person’s height plus a normally distributed random number, with mean zero and variance equal to Vh(1−Vi)/Ve, where Vh is the variance of height. In any analyses involving simulated profile, we used at least a 100 simulations per point of interest.
Estimating proportion of variance explained by a profile and discriminative accuracy (AUC)
The proportion of the variance of sex- and age-adjusted height explained by a profile was estimated using the linear regression model as (1−Vi/Ve), where Vi is the trait’s variance in the model including the profile as a predictor, and Ve is the variance in the model excluding the profile as a predictor.
The receiver-operating characteristic (ROC) curve represents the combinations of sensitivity and specificity for each possible cutoff value of the continuous test result that can be considered to define positive and negative test outcomes. The area under the receiver-operating characteristic curve (AUC) indicates the discriminative accuracy of a continuous test.12 The AUC ranges from 0.5 (total lack of discrimination) to 1.0 (perfect discrimination) and is independent of the prevalence of the condition of interest.13 The AUC can be basically considered as the probability that the test correctly identifies the subject possessing the characteristics of interest (eg, ‘very tall’) from a pair in whom one has and one does not have this characteristic. An AUC of 0.95 means that 95% of the pairs are correctly classified, whereas a test with an AUC of 0.50 is non-discriminative – as accurate as tossing a fair coin.
AUC was computed as the area under the function relating sensitivity to 1–specificity (ROC curve). To derive the ROC curve, we varied the threshold determining ‘positive test result’ from a minimal to a maximal possible test (profile) value. At a given threshold, sensitivity was computed as the proportion of people who test positive among those who do indeed possess the characteristic of interest; the specificity was computed as the proportion of those who test negative among those who do not possess the characteristic of interest.
Results
In all analyses, we used sex- and age-adjusted height as an outcome. We have compared the predictive potential of different methods by contrasting the proportion of the explained height variance explained and AUC. The latter measures the accuracy of the model to discriminate between alternative outcomes (in height context, eg, ‘very tall’ or not).
The data from the population-based Rotterdam Study8 (5748 individuals with complete height, sex, age and genomic data) were used to estimate the predictive potential of the genomic method. In the Rotterdam Study, 34 of the 54 SNPs were significantly associated with height at P<0.05. Only for two SNPs the direction of (non-significant) height association inconsistent with that reported by the original studies (Supplementary Table 1). Before estimating the potential of the genomic profile to predict human height, we also tested whether the 54 loci deviated from the within- or between-loci additivity assumption. After correction for multiple testing, we did not find statistically significant evidence for between-loci interactions (all nominal P>0.001). Only one SNP (rs4794665 located in the NOG-RISK region) showed significant deviation from a within-locus additive model after correction for multiple testing (corrected P=0.0006, see Supplementary Table 1).
The genomic profile, based on 54 recently identified loci, was computed as the sum of the number of height-increasing alleles carried by a person, similar to Weedon et al.5 This profile explained 3.8% of the sex- and age-adjusted variation of height in the Rotterdam Study (Figure 2a). We also estimated the upper explanatory limit of the 54-loci allelic profile by defining the profile as a weighted sum of height-increasing alleles, with weights proportional to the effects estimated in our own data using a multivariable model (Supplementary Table 1). Such a weighted genomic profile explained 5.6% of the variation of height in the Rotterdam Study. The mean difference between people having the ‘highest’ and ‘lowest’ 5% of the genetic height score was 4.9 cm (Figure 2a, Table 1) (6.4 cm when using the weighted profile).

Observed sex- and age-adjusted height vs different predictive profiles. (a) Rotterdam Study, prediction with the genomic profile constructed from 54 loci, (b) ERF study, Galtonian prediction using mid-parental height values and (c) Rotterdam Study, a hypothetical profile explaining 80% of height variance. Red lines: mean residual height in people coming from top and bottom 5% of the profile distribution. Blue line: regression of the height residuals onto profile. In (b), green line has slope of 1, deviation of the blue line from the green showing ‘regression towards mediocrity’.
The ability of the genomic profile to predict a very tall (belonging to the upper 5% of the distribution) person was estimated using the AUC – a statistic routinely used to assess the predictive ability of a test in clinical practice.12, 13, 14, 15 The AUC for the 54-loci genomic profile was 65% (68% for the weighted profile; Table 1 and Figure 3a).

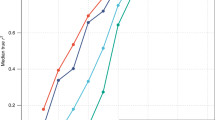
Accuracy to discriminate the top 5% tallest person, as measured by AUC, using different height profiles. (a) 54-loci genomic profile explaining 3.8% (54 loci, solid red line, AUC=65% in the Rotterdam Study), population-specific 54-loci genomic profile explaining 5.8% in the Rotterdam Study (estimated using the data, red dotted line, AUC=68% in the Rotterdam Study), mid-parental value explaining 40% (blue line, AUC=83% in the ERF study) and a hypothetical profile explaining 80% of height variance (green line, AUC=97%). (b) AUC achieved by a test explaining certain proportion of height variance; red: predicting top 50%, blue: predicting top 5%, green: predicting top 1%. Vertical lines: standard error of the mean.
Next, to estimate the predictive power of the Galtonian method, we used the family-based ERF study,9 in which parental height data were available for 550 participants. To construct the Galtonian predictive profile for every person for whom both paternal and maternal heights were available, we computed the average of the parental height residuals. We found that the proportion of height explained by the Galtonian mid-parental profile was 40% (Figure 2b) – which is an order of magnitude higher than the result achieved using the 54-loci genomic profile. The mean difference between people having the ‘highest’ and ‘lowest’ 5% of the mid-parental predictive profile reached an impressive 17.68 cm (Figure 2b, Table 1). Moreover, the Galtonian prediction performed much better when discriminating very tall people (AUC=84%; Table 1 and Figure 3a).
We have addressed the question whether combining the parental height information with genotypic profile leads to better prediction. The analysis was restricted to 270 members of the ERF study for whom both parental phenotype and genetic data were available. Both mid-parental value (P=10−42) and the non-weighted genomic profile (P=0.01) were significantly associated with the height of an offspring. Not surprisingly, the genomic profile was strongly correlated (Pearson’s ρ=0.22, P=0.0003) with the mid-parental height value. Table 1 shows that although statistically significant, considering the genomic profile added little to the prediction based on mid-parental values only (proportion of variance explained increased by ∼1.3%, and AUCs stayed virtually the same).
Finally, we addressed the question of how much variance a genomic profile should explain to achieve a certain AUC value.15 For this, using the Rotterdam Study data, we simulated profiles explaining different proportions of trait variance, and evaluated AUCs for these profiles (Figure 3b). For every evaluated point, one hundred simulations were performed. The simulations have shown that when one aims to predict a person having extreme (1% highest/lowest) value, a predictive profile explaining as little as 17% of the trait’s variance is sufficient to achieve an AUC of 80% (which may generally be considered as good for screening purposes), and a profile explaining 53% to achieve an excellent AUC of 95%. On the other hand, a good prediction of a person from the higher/lower 5% trait’s distribution requires a profile explaining 25%, and an excellent prediction of such a person requires a profile already explaining 68% (Figure 3b).
It can be expected that if all loci controlling human height are known, a genomic profile can explain up to 80% of height variance. Under this scenario, the mean difference between people having the ‘highest’ and ‘lowest’ 5% of such a hypothetical profile was 23.38±0.005 cm (typical realization is presented in Figure 2c). As expected from the high proportion of explained variance, the discriminative accuracy of this hypothetical profile was very high (AUC=97.4±0.3%, Table 1 and Figure 3a).
Discussion
In this work, we compared genomic and Victorian approaches to predict human height. In our data, the 54-loci genomic profile explained 4–6% and Victorian Galton’s mid-parental values explained 40% of the height variance. Adding genomic information to the mid-parental values provided only a small (1.3%) increase in the proportion of variance explained.
In forensics and human medicine, the question of binary classification of a person (eg, ‘very tall’ or not) on the basis of some profile (score) is of high interest. We have proposed earlier that the usefulness of a genomic profile associated with a binary outcome should be evaluated by the area under the ROC curve.14, 15 In medicine, ROC analysis has been extensively used in the evaluation of diagnostic tests. We show that the 54-loci genomic profile had a relatively low discriminative accuracy (AUC=65% for a person falling into 5% tallest). This value, however, is promising for example, approximately the same AUC is reached when predicting the risk of coronary heart disease using low-density lipid levels.16 We estimate that to achieve an AUC of 80% using height genomic profiling, we need to explain at least three times the amount of variance currently explained with the available 54 loci. At the same time, the cheap and straightforward Galtonian approach showed an AUC of 84% when predicting 5% of the tallest person. The latter discriminative accuracy of 84% is better than that of many tests used in the clinical context, such as the Framingham risk scores that predict coronary heart disease based on traditional risk factors such as blood pressure, lipid levels and smoking status.16 However, the Galtonian prediction requires knowledge of parental height, which is not always available in applications such as forensics.
Our height study provides a strong example of a trait for which, at the current stage, a simple prediction based on phenotype of relatives clearly outperforms sophisticated genomic prediction. Would this hold for other phenotypes? The proportion of offspring’s phenotypic variation, which can be explained by a mid-parental phenotypic value, is (h2)2/2, where h2 is the heritability of the trait.17 In a recent study, we have estimated that 11 SNPs explain 3–5% of the variance of total cholesterol, and similar figures were obtained for high- and low-density lipoprotein cholesterol and triglycerides.18 These traits typically exhibit about 30% heritability. Therefore, the Galtonian prediction cannot explain more than 5% of the trait’s variance. Thus, for lipid levels the genomic prediction is already doing as good as (or as bad as) the Galtonian one. However, the genomic profiling, unlike the Galtonian, still has the potential to improve, as more loci affecting the phenotype of interest are discovered.
Although the upper limit for genomic profiling is determined by heritability, the genetic architecture of the trait is a very important factor to consider in estimating the potential of predictive testing.15 For example, for iris color, a single major locus explains the vast proportion of variance, and the AUC of 80% is reached when predicting blue or brown iris color using only three SNPs.19 However, for traits such as blood pressure, only a few loci explaining a very small proportion of variance each are known and, for such traits, the prospects of genomic profiling are much worse.
It can be expected that once all loci involved in human height are shown, the discriminative accuracy of the genomic approach may surpass that of the Galtonian approach. However, it will be a tall order to find all these variants, at least using the current methodology consisting of (meta-analyses) of genome-wide association studies, tailored to capture common variants. The 54 common variants discovered by now, probably already include those with the largest effect sizes. Merely because the variants with the larger effect sizes are most easily captured, the detection of new height genes will require progressively bigger sample sizes (eg, to detect a locus explaining 0.1% of the variance at genome-wide significance P<5 × 10−8 with a power of 80%, one would need to study 40 000 people, whereas to detect a locus explaining 0.01%, one would need 400 000 people).5
As noted by Galton, ‘stature is not a simple element, but a sum of accumulated lengths and thicknesses of more than a hundred of bodily parts… The beautiful regularity in the statures of a population … is due to the number of variable elements the stature is the sum’.1 A detailed analysis of the factors controlling these endophenotypes is likely to be necessary to discover new loci and to make genetic findings useful for applications in forensics and medicine.
We conclude that whereas the genomic approach is potentially more powerful than Victorian Galton's method, the latter will long stay unsurpassed in terms of both discriminative accuracy and costs, when the trait in question is highly heritable and the parental phenotype is usually available. For less heritable traits, such as lipid levels, and in situations when parental information is not available (eg, forensics), genomic methods may provide an alternative, given the variants determining an essential proportion of variation can be identified.
References
Galton F : Regression towards mediocrity in hereditary stature. Journal of the anthropological institute 1886; 15: 246–263.
Fisher RA : The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb 1918; 52: 399–433.
Visscher PM, Medland SE, Ferreira MA et al: Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2006; 2: e41.
Lander ES, Linton LM, Birren B et al: Initial sequencing and analysis of the human genome. Nature 2001; 409: 860–921.
Weedon MN, Lango H, Lindgren CM et al: Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 2008; 40: 575–583.
Lettre G, Jackson AU, Gieger C et al: Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 2008; 40: 584–591.
Gudbjartsson DF, Walters GB, Thorleifsson G et al: Many sequence variants affecting diversity of adult human height. Nat Genet 2008; 40: 609–615.
Hofman A, Breteler MM, van Duijn CM et al: The Rotterdam Study: objectives and design update. Eur J Epidemiol 2007; 22: 819–829.
Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS : The effect of genetic drift in a young genetically isolated population. Ann Hum Genet 2005; 69: 288–295.
Aulchenko YS, Heutink P, Mackay I et al: Linkage disequilibrium in young genetically isolated Dutch population. Eur J Hum Genet 2004; 12: 527–534.
Li Y, Abecasis GR : Rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet 2006; S79: 2290.
Hanley JA, McNeil BJ : The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982; 143: 29–36.
Ash A, Shwartz M : R2: a useful measure of model performance when predicting a dichotomous outcome. Stat Med 1999; 18: 375–384.
Janssens AC, Pardo MC, Steyerberg EW, van Duijn CM : Revisiting the clinical validity of multiplex genetic testing in complex diseases. Am J Hum Genet 2004; 74: 585–588; author reply 588–589.
Janssens AC, Aulchenko YS, Elefante S, Borsboom GJ, Steyerberg EW, van Duijn CM : Predictive testing for complex diseases using multiple genes: fact or fiction? Genet Med 2006; 8: 395–400.
Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB : Prediction of coronary heart disease using risk factor categories. Circulation 1998; 97: 1837–1847.
Falconer DS, Mackay TFC : Introduction to Quantitative Genetics, 4 edn, Harlow, Essex, UK: Addison Wesley Longman, 1996.
Aulchenko YS, Ripatti S, Lindqvist I et al: Genome-wide association study in 16 European population cohorts: major loci influencing lipid levels and coronary heart disease risk. Nat Genet 2009; 41: 47–55.
Kayser M, Liu F, Janssens AC et al: Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am J Hum Genet 2008; 82: 411–423.
Acknowledgements
We thank Professor T Frayling for useful discussion and comments and Dr M Bevova for comments and graphical art support. We acknowledge the work of Dr F Rivadeneira, Dr M Moorhouse, P Arp and M Jhamai, who created the Rotterdam Study genotypic database. This research was supported by grants from the Netherlands Foundation for Scientific Research (NWO), a joint grant from NWO and the Russian Foundation for Basic Research (RFBR), by the Centre for Medical Systems Biology (CMSB) and by the Netherlands Forensic Institute (NFI). MNW is a Vandervell Foundation Research Fellow. Genome-wide genotyping of Rotterdam Study is supported by NWO (175.010.2005.011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Aulchenko, Y., Struchalin, M., Belonogova, N. et al. Predicting human height by Victorian and genomic methods. Eur J Hum Genet 17, 1070–1075 (2009). https://doi.org/10.1038/ejhg.2009.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.5
Keywords
This article is cited by
-
The limits of normal approximation for adult height
European Journal of Human Genetics (2021)
-
Lenz microphthalmia syndrome in neurosurgical practice: a case report and review of the literature
Child's Nervous System (2021)
-
A generalized model for combining dependent SNP-level summary statistics and its extensions to statistics of other levels
Scientific Reports (2019)
-
RETRACTED ARTICLE: Predicting adult height from DNA variants in a European-Asian admixed population
International Journal of Legal Medicine (2019)
-
Using information of relatives in genomic prediction to apply effective stratified medicine
Scientific Reports (2017)
