
Abstract
Isolated cleft lip with or without cleft palate (CL/P) is among the most common human birth defects, with a prevalence of 1 in 700 live births. The paired box (PAX) genes have been suggested as candidate genes for CL/P based largely on mouse models; however, few human studies have focused on this gene family. This study tests for association between markers in four PAX genes and CL/P using a case-parent trio design considering parent-of-origin effects. Trios from four populations (76 from Maryland, 146 from Taiwan, 35 from Singapore, and 40 from Korea) were genotyped for 34 single nucleotide polymorphisms (SNPs) in the PAX3, PAX6, PAX7, and PAX9 genes. We performed the transmission disequilibrium test (TDT) on individual SNPs. Parent-of-origin effects were assessed using the transmission asymmetry test (TAT) and the parent-of-origin likelihood ratio test (PO-LRT). TDT analysis showed one SNP (rs766325) in PAX7 yielding evidence of linkage and association when parent-of-origin was not considered, with an OR(transmission)=1.62 (P=0.003), and five SNPs in PAX6 (including two pairs in near perfect linkage disequilibrium). TAT analysis of all trios revealed two SNPs in PAX7 and four SNPs in PAX3 showing significant excess maternal transmission. For these six SNPs, the maternal OR(transmission) ranged between 1.74 and 2.40, and PO-LRT was also significant (P-values=0.035–0.012). When this analysis was limited to trios with male cases, SNPs in PAX7 showed higher maternal OR(transmission) and greater significance. PAX genes may influence the risk of CL/P through maternal effects, possibly imprinting, which seems to be stronger among male cases.
Similar content being viewed by others

Introduction
Oral clefts are one of the most common birth defects in humans and represent a significant public health burden in terms of both medical and economic burden for affected individuals and their families. Non-syndromic cleft lip with or without cleft palate (CL/P) is complex in its etiology, and both genes and environmental risk factors influence the risk.1 Although several candidate genes have been studied extensively in different populations (TGFA, IRF6, BCL, RARA, etc), relatively few genes have been shown to contain truly causal mutations (MSX1, PVRL1, etc), and these are individually rare and often show incomplete penetrance.2, 3 Recently, several studies have reported that genes responsible for Mendelian malformation syndromes that include CL/P (eg IRF6, which accounts for the majority of Van der Woude syndrome) may also be associated with non-syndromic clefts.2, 4
Paired box (PAX) genes, termed as the PAX gene family, encode for specific DNA-binding transcription factors, which typically contain a PAX domain (an octapeptide) and a paired-type homeodomain.5 The mammalian PAX gene family includes nine genes encoding DNA-binding transcriptional regulatory proteins.6 These nine individual PAX genes are assigned to four subgroups based on conservation of their primary structure: (1) PAX1/PAX9, (2) PAX2/PAX5/PAX8, (3) PAX3/PAX7, and (4) PAX4/PAX6.6
PAX genes play critical roles during fetal development and in the growth of cancer cells. Mutations in the PAX3 (MIM 606597) gene have been associated with Waardenburg syndrome, which can include CL/P.7 PAX3 has also been associated with craniofacial-deafness-hand syndrome.8 PAX7 (MIM 167410) plays a crucial function during neural crest development.9 PAX3 and PAX7 have also been associated with alveolar rhabdomyosarcoma.10 Mutations in PAX6 (MIM 607108) have been associated with aniridia and development of the central nervous system.11, 12 Several studies have reported that PAX genes are associated with CL/P in animals.9, 13 However, to date few studies have focused on PAX genes as risk factors for CL/P in humans.14
It is important to consider parent-of-origin effects when studying birth defects because maternal genotype controls the in utero environment of the developing fetus, and separating maternal genotypic effects from imprinting effects remains an important scientific question.15, 16 Maternal parent-of-origin effects have been suggested for several genes associated with non-syndromic CL/P.17, 18 Males are more often affected with CL/P than females19, 20 however, the underlying cause of this aberrant sex ratio remains unclear. In this paper, we tested for association between single nucleotide polymorphisms (SNPs) in PAX3, PAX6, PAX7, and PAX9 genes and risk of CL/P in 297 case–parent trios, specifically considering parent-of-origin effects in the total sample and stratified by the proband's gender.
Materials and methods
Sample description
As part of an international study of oral clefts, we collected data on case–parent trios recruited through treatment centers in Maryland (MD): Johns Hopkins and University of Maryland; Taiwan (TW): Chang Gung Memorial Hospital; Singapore (SP): KK Women's and Children's Hospital, and Korea (KR): Yonsei Medical Center. Research protocols were reviewed and approved by institutional review boards at each institution. Table 1 lists the gender of all CL/P probands. All parents of probands in the Singaporean, Taiwan, and Korean trios were unaffected, but 4 parents among the 76 MD trios also had an oral cleft. The racial background of case families from MD was 80% European American, 16% African American, and 4% ‘other’. All probands underwent a clinical genetics evaluation (including checking for other congenital anomalies or major developmental delays), and were classified as having an isolated, non-syndromic CL/P. Among the total collection of 297 cases (5% of whom did not specify laterality), 17% of CL cases and 23% of CLP cases were bilateral.
SNP selection, DNA, and genotyping
Single nucleotide polymorphic markers were obtained from literature review and the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/), using a NorthStar Searchlet from Genetic Software Innovations (Cicero, NY, USA), which identified SNPs within each gene based on definitions used in LocusLink and EntrezGene. SNPs were selected with primary consideration given to the spacing between known SNPs and the amount of sequence data available at that time in public databases. SNPs with multiple submitters and higher heterozygosity levels were given priority. SNPs with high ‘design scores’ (a predictor of useable genotypes provided by Illumina, Inc., San Diego, CA, USA), heterozygosity above 0.1 in both Caucasian and Asian populations, and HapMap validation were included. SNPs were selected in and around four PAX genes with the goal of identifying one SNP per 5 kb: 7 SNPs were genotyped for PAX7 on chromosome 1p36.2–p36.12, 13 for PAX3 on chromosome 2q35–q37, 7 for PAX6 on chromosome 11p13, and 7 for PAX9 on chromosome 14q12–q13. A total of 45 SNPs were identified, and 35 were polymorphic in all populations. The call rate we considered acceptable was ≥80%. One SNP had unacceptably high rates of missing data (71%), leaving only 34 SNPs with reasonable heterozygosity for analysis (Table 2).
Genomic DNA samples were prepared from peripheral blood by the protein precipitation method described earlier.21 DNA concentration was determined using the PicoGreen® dsDNA Quantitation Kit (Molecular Probes, Inc., Eugene, OR USA) and all DNA samples were stored at −20°C. A 4-μg aliquot of each genomic DNA sample (concentration of 100 ng/μl) was dispensed into bar-coded 96-well plates and genotyped for SNP markers using the Illumina Golden-Gate chemistry with Sentrix® Array Matrices22 at the SNP Center of the Genetic Resources Core Facility, part of the McKusick–Nathans Institute of Genetic Medicine at the Johns Hopkins School of Medicine. Two duplicates and four CEPH controls were included on each plate to evaluate genotyping consistency within and between plates. Genotypes were generated on a BeadLab 1000 system.23 All SNPs were inspected, and poorly performing SNPs were dropped. No Mendelian inconsistencies were found for these 34 SNPs when checked with the SIB-PAIR program.24
Statistical analysis
Within each population, minor allele frequencies (MAFs) were computed among parents. Pairwise linkage disequilibrium (LD) was computed as r2 for all SNPs using the Haploview program,25 and blocks were identified in Asian and MD population separately (Figure 1). Clayton's extension of the transmission disequilibrium test (TDT) as incorporated into STATA 8.226, 27 was used on individual SNPs to test for evidence of linkage and LD in the total sample of 297 CL/P trios. From this TDT analysis, we calculated the odds ratio of transmission, OR (transmission), and defined a ‘high-risk’ allele as that allele over-transmitted to cases (regardless of its statistical significance).

Linkage disequilibrium as measured by r2 in PAX7, PAX3, PAX6, and PAX9 among parents of CL/P children from Asian and Maryland populations. White: r2=0. Shades of gray: 0<r2<1. Black: r2=1.
Parent-of-origin analyses were conducted in the total sample in several ways. As an initial screening, parent-of-origin effects were examined using the transmission asymmetry test (TAT), suggested by Weinberg et al,28 which is similar to the TDT but excludes mating between two heterozygotes (where transmission can be ambiguous). TAT was stratified into separate paternal and maternal allelic tests. Next, we used the likelihood-based approach proposed by Weinberg29 to confirm these parent-of-origin effects. This log-linear model considers the three mating types where the mother and father carry different numbers of variant alleles, stratified by the number of alleles in the child. This log-linear model is used to compute a parent-of-origin likelihood ratio test (PO-LRT), which tests maternal genotypic effects on the phenotype of the fetus (which could otherwise confound assessment of parent-of-origin effects) along with a separate term for imprinting.29 Here imprinting reflects a differential transmission of alleles to the affected child from mothers versus fathers. PO-LRT was executed using the LEM software.30 We also tested for parent-of-origin effect in the sample stratified by proband’s gender, with separate analyses for trios with male and female cases.
The FAMHAP package was used to estimate haplotype frequencies, while testing for excess transmission of multi-SNP haplotypes.31 In this haplotype analysis, 2–5 SNP haplotypes using sliding window were analyzed using FAMHAP. For this FAMHAP analysis, MD and Asian trios were analyzed separately. The haplotype analysis was carried out ignoring parent-of-origin first. Then the haplotype analysis was conducted for maternal and paternal transmissions separately. FAMHAP calculates maximum likelihood estimates of haplotype frequencies from nuclear families through the expectation–maximization algorithm and is robust in handling missing SNPs.32 This tool provides a haplotype-based test, where the test statistic is based on simulations that randomly permute transmitted and non-transmitted genotypes/haplotypes in each replicate.33 In this analysis, we used max-TDT, which analyzes each haplotype separately and relies on the maximum TDT as the test statistic.33, 34 The program yields P-values corrected for multiple haplotypes.
Results
Among these 34 SNPs, there was considerable variation in allele frequencies among parents from MD and the three Asian populations (Table 2). From the allele frequencies shown in Table 2, it is clear that some markers showed sharp distinctions between MD and Asian samples, whereas others did not. Taiwan, Korean, and Singaporean parents had very similar haplotype frequencies; therefore all Asian trios were combined for haplotype analysis. Patterns of LD across each gene were similar in all populations, with some adjacent SNPs in each gene in perfect LD, rendering them redundant (see Figures 1 and Table 2).
When individual markers were screened with the TDT in the total sample, one SNP in PAX7 and five SNPs in PAX6 were nominally significant when parent-of-origin was ignored (Table 3). The OR(transmission) was 1.62 (P=0.003) for rs766325 in PAX7. The five SNPs in PAX6 showing significant evidence of linkage and LD included two separate pairs of SNPs in perfect LD. The most significant SNP (rs3026354) gave an OR(transmission)=1.47 (P=0.008) ignoring parent-of-origin (Table 3). When analyzed separately in each of the four populations, the association was less strong because of the smaller sample sizes, but the patterns of OR(transmission) were similar (data not shown).
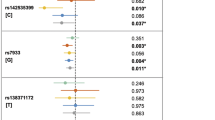
Parent-of-origin effects were first investigated by stratifying informative transmissions and non-transmissions by parental source for all SNPs in the total dataset (Table 4). TAT (ie where heterozygous × heterozygous matings were dropped) revealed three SNPs showing excess maternal transmission significant at the P<0.01 level (rs618941, rs553934 in PAX7, and rs1367414 in PAX3; see Figure 2 and Table 4), and three others (rs4674639, rs930140, and rs7600206) in PAX3 showing slightly less significant transmission from mothers. For these six SNPs, estimated maternal OR(transmission) was statistically significant (ranging from 1.74 to 2.40) in TAT analysis. The PO-LRTs were also significant for these six SNPs (P-values ranging from 0.035 to 0.012) and gave estimated risk ratios for imprinting ranging between 2.08 and 2.78 for these six SNPs, suggesting excess maternal transmission of this region in PAX7 and PAX3 (Table 4). Parent-of-origin effects for markers in PAX6 and PAX9 were not significant (data not shown).

Empirical P-values for individual SNPs from PAX7, PAX3, PAX6, and PAX9 genes in 297 CL/P case–parent trios from four populations (Maryland, Singapore, Taiwan, and Korea) combined. (a) Only maternal transmission was considered; (b) only paternal transmission was considered.
Separate analyses were conducted for trios with male and female cases. For two SNPs in PAX7, the estimated OR(transmission) from mothers to male case was statistically significant (OR=4.50, P=0.0003 for rs553934; and OR=4.20, P=0.0017 for rs618941). Both of these SNPs gave significant PO-LRTs (P=0.028 for rs618941 and P=0.027 for rs553934). Among trios with a female case, however, OR(transmission) and PO-LRT were non-significant for these two SNPs (data not shown).
In the haplotype analysis using sliding window (ignoring parent-of-origin), haplotypes of two SNPs (rs766325 and rs880810) in PAX7 showed evidence of excess transmission of the 1–2 haplotype to the case among Asian trios (P=0.036). MD trios showed similar transmission patterns, but were not statistically significant (data not shown). In PAX6, haplotypes of three SNPs (rs592859, rs3026354, and rs2071164) showed strong evidence of excess transmission of the 1–2–2 haplotype to the case among Asian trios (P=0.011). The MD trios again showed similar transmission patterns, but were not statistically significant (data not shown). Haplotypes in PAX3 and PAX9 genes were not significant at P-value=0.05 (data not shown).
Next, we conducted the haplotype analyses stratified by parent-of-origin using 2–5 sliding windows (Table 5). In PAX7, haplotypes of rs880810 and rs618941 were most significant. The 2–2 haplotype showed evidence of excess maternal transmission to the CL/P child among Asian trios (P=0.049) and among MD trios (P=0.041), whereas no haplotypes showed deviation from expected when inherited from fathers. Analysis of two SNPs in PAX3 (rs4674639 and rs930140) showed evidence of excess maternal transmission of the 1–2 haplotype, with stronger evidence coming from Asian trios (here again MD trios showed similar but non-significant patterns of over-transmission).
Discussion
Our study of case–parent trios from different populations (comprising a total of 297 CL/P trios) showed evidence of linkage in the presence of LD for multiple SNPs in the PAX7 and PAX3 genes only when parent-of-origin effects were considered. In this study, ignoring parent-of-origin made the PAX7 and PAX3 genes look relatively uninteresting. Only a single SNP in PAX7 showed any evidence of linkage and LD. However, considering parent-of-origin revealed two SNPs in PAX7 and three SNPs in PAX3 yielding strong evidence of linkage and LD when transmitted from mothers but not from fathers. This evidence was more dramatic among male cases. Other studies also report that ignoring parent-of-origin could lead to overlooking important genes. In a case–parent trio study for bipolar I disorder, TDT analysis revealed no statistically significant association with SNPs on chromosome 18p11. However, when parent-of-origin was considered, evidence of association was seen involving two potentially causal loci.35
In screening for parent-of-origin effects, we found suggestive evidence of excess maternal transmission for several SNPs in PAX3 and PAX7, which are closely related and are important in mammalian embryogenesis.36 Relaix et al37 identified a new cell population expressing transcription factors PAX3 and PAX7, but no skeletal muscle-specific markers. These cells are maintained as a proliferating cell line throughout development in embryonic and fetal muscles of the trunk and limbs.
Excess maternal transmission could reflect genomic imprinting or direct maternal genotype effects on the developing fetus. Maternal genotypic effects for non-syndromic CL/P have also been reported for several other candidate genes (MTHFR, CBS, and TGFB3), but these are yet to be confirmed.17, 18, 38
In this study, log-linear models discriminating between maternal genotype and child genotype effects revealed a possible maternal imprinting effect for multiple SNPs in PAX7 and PAX3. Estimates of maternal genotype effects were generally non-significant for the 19 SNPs in PAX7 and PAX3, except for a single SNP (rs1367414). Genomic imprinting is defined as the differential expression of alleles depending on parent-of-origin.39 A common feature of imprinted genes is DNA sequence carrying a gametic methylation imprint, known as gametic DMR (Differentially DNA-Methylated Region).40 Parental allele-specific DNA methylation has been found at most imprinted clusters examined thus far. For example, the IGF2 cluster has a gametic DMR located 2 kb upstream from the H19 nc RNA promoter, which is methylated only in the paternal gamete and is maintained thereafter in all somatic tissues.41 Kurmasheva et al42 suggested PAX3 gene methylation may be correlated with gene inactivation.
In a variety of animal species, maternal transcripts and proteins control early embryonic development in the developing oocyte.43 In the leech Helobdella, Woodruff et al44 found that Hau-Pax3/7A is present as a maternal transcript in both ectodermal and mesodermal progenitor cells. They suggested Hau-Pax3/7A plays an important role in mesoderm development. Helobdella embryos receive a large contribution of maternal Hau-Pax3/7A RNA, but its function remains unknown.44
Many congenital anomalies occur more often in one gender. Males are more often affected with CL/P than females.19, 20 Rittler et al20 reported that infants with CL/P were more frequently female when the father was older, and among CL cases, this shift in sex ratio was highly significant. In our results from markers in PAX7 and PAX3 genes, boys showed stronger evidence of possible imprinting than female cases.
Even though this candidate gene study involved a modest number of SNPs in each gene, addressing the issue of multiple comparisons is necessary before an overall statement about the significance of our findings can be made. Here we relied on a hypothesis-driven approach for single SNP analysis and haplotype-based test statistics. SNPs in strong LD typically have highly correlated P-values, adjusting significance levels through Bonferroni correction is overly conservative. Therefore, following the strategy in Sull et al,45 we adjusted empirical P-values for the number of LD blocks rather than the number of SNPs. In this study, we have 10 LD blocks in these four genes (three forPAX7, three for PAX3, two for PAX6, and two for PAX9). In the second block with two SNPs in PAX7 gene (as shown in Table 4), we found evidence against the null hypothesis only for maternal transmission (the empirical P-value of 0.006 would still be marginally significant after correcting for the number of LD blocks). We also used haplotype-based test statistics based on permutation analysis of case–parent trio data. Salyakina et al46 argue that permutation tests are generally preferred over adjustments of asymptotic P-values based on the estimated correlation structure among multiple markers or on conventional Bonferroni adjustment (which can be too conservative).47 The case–parent trio design offers the advantage of testing directly for maternal versus paternal effects, and allows separating these from effects of the fetal genotype versus parental origin in a robust manner.26, 48 Another advantage of this design is that it minimizes confounding that plagues traditional case–control designs. This permits pooling trios from four diverse populations into a combined test of allelic effects on OR(transmission), while testing for parent-of-origin effects. The present study showed excess maternal transmission of markers in PAX7 and PAX3, suggesting that these genes may influence the risk of CL/P, possibly through imprinting. Independent confirmation is still needed to determine the ultimate impact of these PAX genes on risk to CL/P.
References
Cobourne MT : The complex genetics of cleft lip and palate. Eur J Orthod 2004; 26: 7–16.
Zucchero TM, Cooper ME, Maher BS et al: Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med 2004; 351: 769–780.
Carinci F, Scapoli L, Palmieri A, Zollino I, Pezzetti F : Human genetic factors in nonsyndromic cleft lip and palate: an update. Int J Pediatr Otorhinolaryngol 2007; 71: 1509–1519.
Stanier P, Moore GE : Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet 2004; 13: R73–R81.
Volcik KA, Blanton SH, Kruzel MC et al: Testing for genetic associations with the PAX gene family in a spina bifida population. Am J Med Genet 2002; 110: 195–202.
Underhill DA : Genetic and biochemical diversity in the Pax gene family. Biochem Cell Biol 2000; 78: 629–638.
Tassabehji M, Read AP, Newton VE et al: Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat Genet 1993; 3: 26–30.
Asher Jr JH, Sommer A, Morell R, Friedman TB : Missense mutation in the paired domain of PAX3 causes craniofacial-deafness-hand syndrome. Hum Mutat 1996; 7: 30–35.
Basch ML, Bronner-Fraser M, Garcia-Castro MI : Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 2006; 441: 218–222.
Barr FG, Nauta LE, Davis RJ, Schafer BW, Nycum LM, Biegel JA : In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet 1996; 5: 15–21.
Chao LY, Mishra R, Strong LC, Saunders GF : Missense mutations in the DNA-binding region and termination codon in PAX6. Hum Mutat 2003; 21: 138–145.
Heins N, Malatesta P, Cecconi F et al: Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci 2002; 5: 308–315.
Peters H, Neubuser A, Kratochwil K, Balling R : Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 1998; 12: 2735–2747.
Ichikawa E, Watanabe A, Nakano Y et al: PAX9 and TGFB3 are linked to susceptibility to nonsyndromic cleft lip with or without cleft palate in the Japanese: population-based and family-based candidate gene analyses. J Hum Genet 2006; 51: 38–46.
Weinberg CR, Umbach DM : A hybrid design for studying genetic influences on risk of diseases with onset early in life. Am J Hum Genet 2005; 77: 627–636.
Wilkins JF, Haig D : What good is genomic imprinting: the function of parent-specific gene expression. Nat Rev Genet 2003; 4: 359–368.
van Rooij IA, Vermeij-Keers C, Kluijtmans LA et al: Does the interaction between maternal folate intake and the methylenetetrahydrofolate reductase polymorphisms affect the risk of cleft lip with or without cleft palate? Am J Epidemiol 2003; 157: 583–591.
Rubini M, Brusati R, Garattini G et al: Cystathionine beta-synthase c.844ins68 gene variant and non-syndromic cleft lip and palate. Am J Med Genet 2005; 136: 368–372.
Niswander JD, MacLean CJ, Chung CS, Dronamraju K : Sex ratio and cleft lip with or without cleft palate. Lancet 1972; 2: 858–860.
Rittler M, Lopez-Camelo J, Castilla EE : Sex ratio and associated risk factors for 50 congenital anomaly types: clues for causal heterogeneity. Birth Defects Res A Clin Mol Teratol 2004; 70: 13–19.
Bellus GA, Hefferon TW, Ortiz de Luna RI et al: Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 1995; 56: 368–373.
Oliphant A, Barker DL, Stuenlpnagel JR, Chee MS : BeadArray™ Technology: enabling an accurate, cost-efficient approach to high-throughput genotyping. Biotechniques 2002; 32: S56–S61.
Fan JB, Oliphant A, Shen R et al: Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 2003; 68: 69–78.
Duffy DL : Sib-pair, Version 1.00a. Brisbane: Queensland Institute of Medical Research, 2007.
Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Spielman RS, McGinnis RE, Ewens WJ : Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993; 52: 506–516.
Cordell HJ, Barratt BJ, Clayton DG : Case/pseudocontrol analysis in genetic association studies: a unified framework for detection of genotype and haplotype associations, gene-gene and gene-environment interactions, and parent-of-origin effects. Genet Epidemiol 2004; 26: 67–185.
Weinberg CR, Wilcox AJ, Lie RT : A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet 1998; 62: 969–978.
Weinberg CR : Methods for detection of parent-of-origin effects in genetic studies of case-parents triads. Am J Hum Genet 1999; 65: 229–235.
van Den Oord EJ, Vermunt JK : Testing for linkage disequilibrium, maternal effects, and imprinting with (in)complete case-parent triads, by use of the computer program LEM. Am J Hum Genet 2000; 66: 335–338.
Becker T, Baur MP, Knapp M : Detection of parent-of-origin effects in nuclear families using haplotype analysis. Hum Hered 2006; 62: 64–76.
Becker T, Knapp M : Maximum-likelihood estimation of haplotype frequencies in nuclear families. Genet Epidemiol 2004; 27: 21–32.
Zhao H, Zhang S, Merikangas KR et al: Transmission/disequilibrium tests using multiple tightly linked markers. Am J Hum Genet 2000; 67: 936–946.
Schaid DJ : General score tests for associations of genetic markers with disease using cases and their parents. Genet Epidemiol 1996; 13: 423–449.
Mulle JG, Fallin MD, Lasseter VK, McGrath JA, Wolyniec PS, Pulver AE : Dense SNP association study for bipolar I disorder on chromosome 18p11 suggests two loci with excess paternal transmission. Mol Psychiatry 2007; 12: 367–375.
Mansouri A, Hallonet M, Gruss P : Pax genes and their roles in cell differentiation and development. Curr Opin Cell Biol 1996; 8: 851–857.
Relaix F, Rocancourt D, Mansouri A, Buckingham M : A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005; 435: 948–953.
Reutter H, Birnbaum S, Mende M et al: TGFB3 displays parent-of-origin effects among central Europeans with nonsyndromic cleft lip and palate. J Hum Genet 2008; 53: 656–661.
Iwasa Y : The conflict theory of genomic imprinting: how much can be explained? Curr Top Dev Biol 1998; 40: 255–293.
Parham DM, Ellison DA : Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med 2006; 130: 1454–1465.
Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA : Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993; 362: 751–755.
Kurmasheva RT, Peterson CA, Parham DM, Chen B, McDonald RE, Cooney CA : Upstream CpG island methylation of the PAX3 gene in human rhabdomyosarcomas. Pediatr Blood Cancer 2005; 44: 328–337.
Davidson EH : Gene Activity in Early Development. Orlando, FL: Academic Press, 1986.
Woodruff JB, Mitchell BJ, Shankland M : Hau-Pax3/7A is an early marker of leech mesoderm involved in segmental morphogenesis, nephridial development, and body cavity formation. Dev Biol 2007; 306: 824–837.
Sull JW, Liang KY, Hetmanski JB et al: Differential parental transmission of markers in RUNX2 among cleft case-parent trios from four populations. Genet Epidemiol 2008; 32: 505–512.
Salyakina D, Seaman SR, Browning BL, Dudbridge F, Muller-Myhsok B : Evaluation of Nyholt's procedure for multiple testing correction. Hum Hered 2005; 60: 19–25.
Nyholt DR : A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet 2004; 74: 765–769.
Starr JR, Hsu L, Schwartz SM : Assessing maternal genetic associations: a comparison of the log-linear approach to case-parent triad data and a case-control approach. Epidemiology 2005; 16: 294–303.
Acknowledgements
This research was supported by R21-DE-013707 and R01-DE-014581 from the National Institute of Dental & Craniofacial Research and Korean Research Foundation (2004-041-E00104), and the Seoul City R&BD program (10526) in Korea. We thank all participants who donated samples for this multi-center study of oral clefts, as well as the staff at each participating site and institution. We also thank Gerald Raymond for his assistance in screening CL/P patients at Hopkins.
Author information
Authors and Affiliations
Corresponding author
Additional information
Web Resources
Online Mendelian Inheritance in Man (OMIM): http://www.ncbi.nlm.nih.gov/Omim/
Rights and permissions
About this article
Cite this article
Sull, J., Liang, KY., Hetmanski, J. et al. Maternal transmission effects of the PAX genes among cleft case–parent trios from four populations. Eur J Hum Genet 17, 831–839 (2009). https://doi.org/10.1038/ejhg.2008.250
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.250
Keywords
This article is cited by
-
Rare variant modifier analysis identifies variants in SEC24D associated with orofacial cleft subtypes
Human Genetics (2023)
-
PAX7 gene polymorphism in muscular temporomandibular disorders as potentially related to muscle stem cells
BMC Musculoskeletal Disorders (2021)
-
Genome-wide analysis of parent-of-origin effects in non-syndromic orofacial clefts
European Journal of Human Genetics (2014)
-
Male and female differential reproductive rate could explain parental transmission asymmetry of mutation origin in Hirschsprung disease
European Journal of Human Genetics (2012)
-
Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci
Nature Genetics (2012)
